
小反刍兽疫病毒融合蛋白基因的克隆、序列分析及表达
2011-10-14 02:54倪伟才学鹏乔军孟庆玲陈创夫任艳
石河子大学学报(自然科学版) 2011年1期
倪伟,才学鹏,乔军,孟庆玲,陈创夫,任艳
(1石河子大学动物科技学院预防兽医学重点实验室,石河子832003;
2中国农业科学院兰州兽医研究所家畜疫病病原生物学国家重点实验室,兰州730046)
小反刍兽疫病毒融合蛋白基因的克隆、序列分析及表达
倪伟1,2,才学鹏2,乔军1,孟庆玲1,2,陈创夫1,任艳1
(1石河子大学动物科技学院预防兽医学重点实验室,石河子832003;
2中国农业科学院兰州兽医研究所家畜疫病病原生物学国家重点实验室,兰州730046)
根据 GenBank报道的小反刍兽疫病毒(PPRV)融合蛋白(F)基因序列,用特异性引物对 PPRV疫苗株F蛋白基因进行了RT-PCR扩增,并将其克隆到p GEM-T载体中进行测序。结果表明:F基因ORF全长1641 bp,编码546个氨基酸;推导的氨基酸序列中第1~18位氨基酸构成信号肽序列,第488~510氨基酸为跨膜区。构建原核表达载体pETF1和pETF2,转化 E.coliBL21(DE3),用 IPTG诱导表达。SDS-PAGE和Western-blotting的分析结果表明,F1和F2基因在大肠杆菌中均获得了表达,且均具有良好的反应原性。用Ni-NTA试剂盒纯化F1和F2重组蛋白,为研发检测PPRV特异性抗体的诊断试剂奠定了基础。
小反刍兽疫病毒;融合蛋白基因;克隆;原核表达
小反刍兽疫是由小反刍兽疫病毒(Peste des petits ruminantsvirus,PPRV)引起的一种急性、高度接触性传染病[1-2]。该病严重危害山羊和绵羊等反刍动物,其发病率可达80%~100%,严重暴发期时的病死率为100%,被世界动物卫生组织列为A类动物疫病,在我国被列为一类动物传染病。近年来,该病流行范围有不断扩大的趋势[3-4]。因此,有必要加强我国边境地区动物血清学检测和监测。
目前,虽然国内外已经建立了诊断该病的琼脂糖凝胶免疫扩散、对流免疫电泳、间接荧光抗体试验、病毒微量中和试验、免疫捕获ELISA等,但这些方法或操作复杂费时或灵敏度、特异性不高或没有商品化的试剂盒,这给基层单位PPR流行病学调查与监测带来了较大困难。鉴于此,建立一种特异、敏感、快速检测方法势在必行。
由于我国过去一直属于小反刍兽疫非疫区国家,因此缺乏对于该病的诊断及监测试剂的研发,这给该病的防控带来困难。融合蛋白(F)是PPRV的主要结构蛋白之一,位于 PPRV病毒囊膜的表面,它既是PPRV感染宿主细胞的关键蛋白也是诱导机体产生中和抗体的保护性抗原[5-6]。为了研制PPRV特异性检测用抗原,本研究采用基因工程手段表达PPRV F蛋白,以期为建立检测PPRV特异性抗体的诊断试剂奠定基础。
1 材料与方法
1.1 细胞和载体
大肠杆菌感受态细胞 E.coliJM109、E.coli BL21(DE3)和表达载体pET-28a(+)均为本室保存。p GEM-T载体试剂盒购自Promega公司。
1.2 主要试剂
Taq plus DNA 聚合酶、DNA Marker、辣根过氧化物酶标记兔抗山羊 HRP-IgG,购自北京天根生化科技有限公司。DNA回收试剂盒、AMV反转录酶、RNA酶抑制剂、限制性内切酶BamHⅠ、XhoⅠ、HindⅢ,购自 Promega公司。Ni-NTA蛋白纯化试剂盒购自Qiagen公司。PPRV疫苗株总RNA和山羊抗PPRV阳性血清由中国农业科学院兰州兽医研究所惠赠。
1.3 PPRV F蛋白基因的RT-PCR扩增与克隆
RT反应:20μL反应体系。取 PPRV疫苗株总RNA加入AMV 5×buffer 4μL、RNA酶抑制剂1μL 、2.5 mmol/L dNTP 4μL 、OligodT 1μL 、10 U/μL AMV 反转录酶 5 U、ddH2O 9.5μL,于 42 ℃反应1 h。
PCR扩增:根据 GenBank中已有的 PPRV全基因组序列(登录号:AJ849636),用 Primer 5.0软件设计P1和P2引物:

PCR采用50μL反应体系,体系组成为:10×反应缓冲液(含 15 mmol/L MgCl2)5μL,dNTP(2.5 mmol/L)4μL,上游引物 P1(25μmol/L)1 μL,下游引物 P2(25μmol/L)1μL,模板(反转录的cDNA)10μL,Taq plus酶 1.5 U,补水至 50μL。
PCR反应条件:96℃200 s;94℃45 s,58℃45 s,72℃2 min,35个循环;最后72℃延伸10 min。
PCR产物用1.5%琼脂糖凝胶电泳分析。用DNA回收试剂盒回收PCR产物,与p GEM-T载体4℃过夜连接,次日转化 E.coliJM109感受态细胞,在氨苄抗性的LB固体培养基上37℃过夜培养,用蓝白斑和 PCR方法筛选阳性克隆(命名为p TF),随机选取3个鉴定正确的阳性克隆送上海生物工程有限责任公司进行序列测序。
1.4 原核表达载体pETF1和pETF2的构建
根据测定的F基因序列,分析其推导的氨基酸序列,选择 F1(第438~911位核苷酸)和 F2(第 921~1388位核苷酸)编码抗原表位集中的区域,分别用P3-P4和P5-P6引物,以pTF为模板进行扩增。
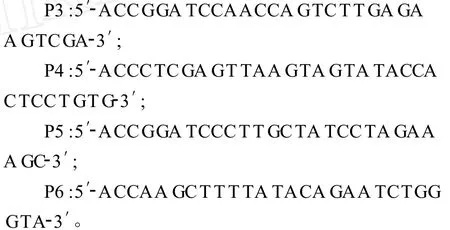
分别回收 F1和 F2基因,用限制性内切酶BamHⅠ、XhoⅠ对 F1基因进行双酶切,用限制性内切酶BamHⅠ、HindⅢ对F2基因进行双酶切,然后将酶切后的F1和F2片段分别于用相应限制性内切酶处理的pET-28a(+)载体连接,分别用两种连接产物转化 E.coliBL21(DE3)感受态细胞。转化后的BL21细胞涂于含卡那霉素的LB固体培养基上,37℃过夜培养。挑取单菌落进行摇菌,提取质粒,进行PCR和双酶切鉴定。
1.5 F1和F2基因的诱导表达及纯化
将鉴定正确的重组载体转化 E.coliBL21(DE3)表达菌,37℃振荡培养至OD600值约为0.6,加入终浓度为1.5 mmol/L IPTG进行诱导表达,并进行 SDS-PA GE电泳分析。将SDS-PAGE电泳凝胶电转至硝酸纤维薄膜上,以山羊抗 PPRV阳性血清为一抗,HRP标记的兔抗山羊 IgG为二抗,进行Western-blotting分析。用Ni-N TA蛋白纯化试剂盒,按照试剂盒说明书进行 F1和 F2蛋白的纯化,将最后的洗脱液进行SDS-PA GE电泳检测。
2 结果与分析
2.1 PPRV F基因的扩增与克隆结果
经1.5%琼脂糖凝胶电泳分析的结果(图1)表明,PCR扩增产物大小约为1650 bp,与预期大小相符。图2显示,以重组载体p TF为模板,可以扩增与与预期大小一致的目的基因片段;限制性内切酶EcoRⅠ、XhoI和 HindIII酶切p TF,酶切片段大小与预期结果一致。
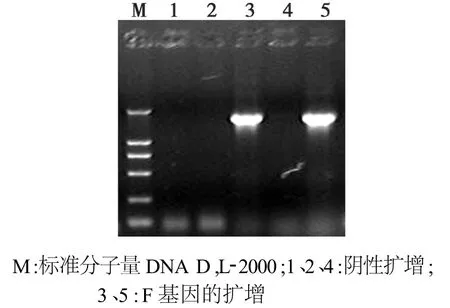
图1 PPRV F基因的扩增Fig.1 Amplification of F gene of PPRV

图2 重组载体pTF的酶切鉴定Fig.2 Identification of pTF by restriction endonuclease digestion
2.2 PPRV F基因的测序及序列分析
1641 bp,编码546个氨基酸(图3)。
经测序,PPRV疫苗株F蛋白cDNA ORF全长

图3 PPRV F蛋白基因cDNA序列及其推导的氨基酸序列Fig.3 cDNA sequence of F gene of PPRV and it’s deduced amino acid sequence
将图3与 GenBank登录的 Turkey 2000毒株相比,F基因存在2个不同的变异位点(第222位A→T,第963位核苷酸G→T),但均为同义突变。运用Signal IP和TMHMM软件分析发现,推导的氨基酸序列中第1~18位氨基酸构成信号肽序列,第488~510位氨基酸残基为跨膜区。Prosite软件分析发现,推导的F蛋白上有3个潜在的N-联糖基化位点(25~28、57~60、63~66)、9 个蛋白激酶C磷酸化位点(38~40、106~108、145~147、216~218、358~360、372~374、413~415、482~484、538~540)、9 个酪蛋白激酶 II磷酸化位点(59~62、65~68、148~151、216~219、248~251、266~269、283~286、323~326、464~467)、2 个酪氨酸激酶磷酸化位点(240~247、431~438)、15个N-豆蔻酰化位点(30~35、33~38、111~116、116~121、120~125、235~240、336~341、352~362、369~374、380~385、456~461、460~465、485~490、501~506、537~542),1 个酰胺化位点(102~105)。氨基酸序列分析结果提示,F蛋白需要经过糖基化和磷酸化后才能发挥其生物学功能。
2.3 原核表达载体pETF1和pETF2鉴定结果
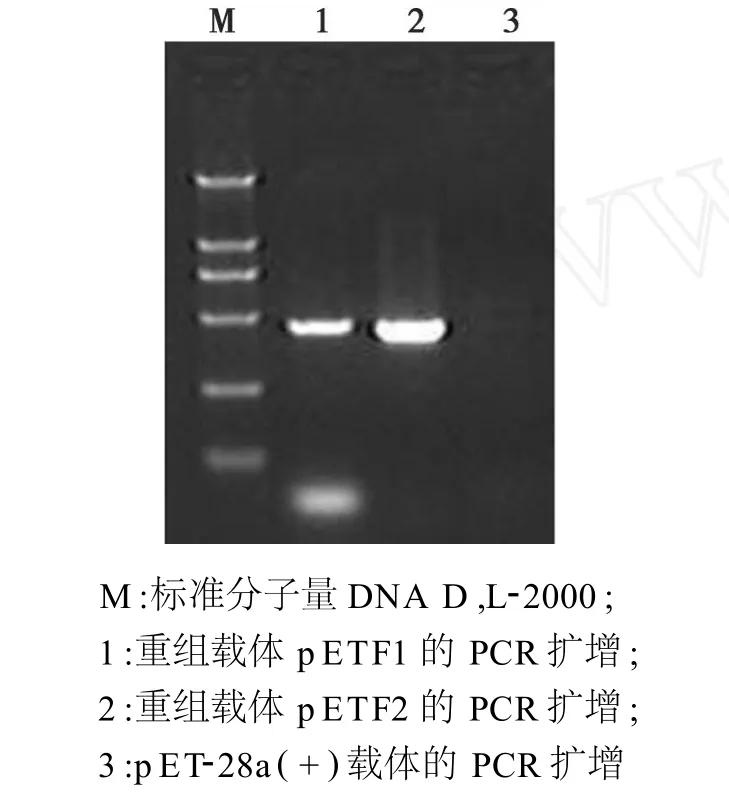
图4 重组载体pETF1和pETF2的PCR鉴定Fig.4 Identification of recombinant pETF1 and pETF2 by PCR
以pETF1和pETF2为模板,分别用 P3-P4和P5-P6引物对其进行扩增,结果扩增的基因片段与F1和F2基因大小相符,分别为473 bp和468 bp(图4)。用限制性内切酶双酶切鉴定,结果得到了与预期一致的结果。
2.4 F1和 F2基因的表达及Western-blotting分析
将转化pETF1和pETF2质粒的 E.coliBL21(DE3)重组菌用IPTG诱导培养,SDS-PAGE电泳分析发现在诱导后出现了2条相对分子量相同(约20.6 kDa)的蛋白带,与预期的蛋白的相对分子量一致(图5中的第6~8道)。以山羊抗PPRV阳性血清为一抗,兔抗山羊 HRP-IgG为二抗,Western-blotting分析发现,表达的F1和F2重组蛋白均能与阳性血清发生反应,提示F1和F2重组蛋白具有良好的反应原性。
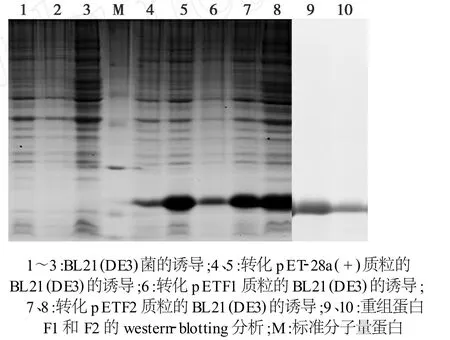
图5 PPRV F1和F2基因的表达及Western-blotting分析Fig.5 Analysis of F1 and F2 recombinant proteins by SDS-PAGE and Western-blotting
2.5 F1和F2蛋白的纯化结果
将纯化的F1和F2重组蛋白进行SDS-PAGE电泳检测,在约20.6 kDa处可见纯化的 F1和 F2蛋白条带(图6)。

图6 PPRV F1、F2重组蛋白纯化结果Fig.6 Purification of recombinant F1 and F2 protein by Ni-NTA Agarose
3 讨论
自从1942年首次报道 PPR以来,全球目前至少已经有29个国家暴发了该病[1-2,13-15],2007年7月我国西藏自治区日土县热帮乡龙门卡村山羊首次爆发PPRV感染,标志着该病已经传入我国。由于我国过去一直属于PPR非疫区国家,因此国内有关PPR的流行病学资料几乎是空白。同时,与国外相比在 PPR的检测和监测技术研究上也相对滞后[8-12],这给国内PPR流行病学调查与监测工作带来了很大的困难。新疆作为我国最大的以牛羊养殖业为主的西部边疆省份,其地貌复杂,兽医防疫体系相对较弱,并且与8个国家接壤,边境线全长5600 km,而与新疆接壤的印度、尼泊尔、巴基斯坦、哈萨克斯坦、塔吉克斯坦等国家都曾大规模暴发过PPR疫情,因而 PPR已经严重威胁到新疆畜牧业的安全。因此,严密监测和防范PPR传入我国已刻不容缓。
PPRV基因组全长15948核苷酸(Turkey 2000),编码6种结构蛋白和2种非结构蛋白[5,10],即核蛋白(N)、磷蛋白(P)、多聚酶大蛋白(L)、基质蛋白(M)、融合蛋白(F)和血凝素蛋白(H),其中 F蛋白是PPRV侵染宿主细胞的关键蛋白之一[6-7],同时也是诱导机体产生中和抗体的主要保护性抗原,因此克隆和表达F基因对于PPR的免疫预防制剂和诊断试剂的研制具有重要的意义。在本研究中,我们首先对PPRV疫苗株F全长基因进行了克隆和序列分析,然后运用DNAstar软件对F蛋白的抗原表位进行了分析,发现F1(第438~911位核苷酸)和 F2区域(第 921~1390位核苷酸)编码的抗原表位比较集中,因此将这2个基因片段进一步亚克隆入pET-28a表达载体中进行诱导表达。Western-blotting检测的结果表明,表达的重组 F1和 F2蛋白均具有良好的反应原性;同时,利用镍离子柱制备的纯度较高的重组F1和F2蛋白,这为进一步研发特异性强的PPRV诊断试剂奠定了良好的基础。
[1]Tufan M.Animal health authorities and transboundary animal diseases in Turkey[J].J Vet Med B Infect Dis Vet Public Health,2006,53(1):35-37.
[2]Kwiatek O,Minet C,Grillet C,et al.Peste des petits ruminants(PPR)outbreak in Tajikistan[J].JComp Pathol,2007,136(3):111-119.
[3]Abraham G,Sintayehu A,Libeau G,et al.Antibody seroprevalences against peste des petits ruminants(PPR)virus in camels,cattle,goats and sheep in Ethiopia[J].Prev Vet Med,2005,70(1/2):51-57.
[4]Osman N A,Ali A S,Rahman M E,et al.Antibody seroprevalences against Peste des Petits Ruminants(PPR)virus in sheep and goats in Sudan[J].Trop Anim Health Prod,2009,41(7):1449-1453.
[5]Bailey D,Banyard A,Dash P,et al.Full genome sequence of peste des petits ruminants virus,a member of the Morbillivirus genus[J].Virus Research,2005,110:119-124.
[6]Dhar P,Muthuchelvan D,Sanyal A,et al.Sequence analysis of the haemagglutinin and fusion protein genes of peste-des-petits ruminants vaccine virus of Indian origin[J].Virus Genes,2006,32(1):71-78.
[7]Kerur N,Jhala M K,Joshi C G.Genetic characterization of Indian peste des petits ruminants virus(PPRV)by sequencing and phylogenetic analysis of fusion protein and nucleoprotein gene segments[J].Res Vet Sci,2007,26(10):456-463.
[8]Balamurugan V,Singh R P,Saravanan P,et al.Development of an indirect ELISA for the detection of antibodies against peste-des-petits-ruminants virus in small ruminants[J].Vet Res Commun,2007,31(3):355-364.
[9]George A,Dhar P,Sreenivasa B P,et al.The M and N genes-based simplex and multiplex PCRs are better than the F or H gene-based simplex PCR for Peste-des-petits-ruminants virus[J].Acta Virol,2006,50(4):217-222.
[10]Muthuchelvan D,Sanyal A,Balamurugan V,et al.Sequence analysis of the nucleoprotein gene of Asian lineage peste des petits ruminants vaccine virus[J].Vet Res Commun,2006,30(8):957-963.
[11]Couacy Hymann E,Bodjo S C,Danho T,et al.Early detection of viral excretion from experimentally infected goats with peste-des-petits ruminants virus[J].Prev Vet Med,2007,78(1):85-88.
[12]Sreenivasa B P,Singh R P,Mondal B,et al.Marmoset B95a cells:a sensitive system for cultivation of Peste des petits ruminants(PPR)virus[J].Vet Res Commun,2006,30(1):103-108.
[13]Yesilbaˇg K,Yilmaz Z,GölcüE,et al.Peste des petits ruminants outbreak in western Turkey[J].Veterinary Record,2005,157(9):260-261.
[14]Abraham G,Sintayehu A,Libeau G,et al.Antibody seroprevalences against peste des petits ruminants(PPR)virus in camels,cattle,goats and sheep in Ethiopia[J].Prev Vet Med,2005,70(1/2):51-57.
[15]Frölich K,Hamblin C,Jung S,et al.Serologic surveillance for selected viral agents in captive and free-ranging populations of Arabian oryx (Oryx leucoryx)from Saudi Arabia and the United Arab Emirates[J].J Wildl Dis,2005,41(1):67-79.
Cloning,Sequence Analysis and Expression of Fusion Gene
ofPeste des petits ruminantsVirus
NI Wei1,CAI Xuepeng2,QIAO Jun1,MENG Qingling1,CHEN Chuangfu1,REN Yan1
(1 The Key Lab of Preventive Veterinary Science,College of Animal Science and Technology,Shihezi University,Shihezi 832003,China;2 State Key Lab of Veterinary Etiological Biology,Lanzhou Veterinary Research Institute,Chinese Academy of Agricultural Science,Lanzhou 730046,China)
According to fusion(F)protein gene sequence ofPeste des petits ruminantsvirus strain reported by GenBank,a pair of specific primers was designed and used to amplify F gene of PPRV vaccinal strain.The RT-PCR product was purified and ligatured with p GEM-T for sequencing.The open reading frame length of F gene of PPRV strain was 1641 bp,which encoded 546 amino acids.The front 18 amino acids constitute signal peptide,and 488-510 amino acids form transmembrane.Then F1 and F2 genes were subcloned into pET-28a(+)to generate prokaryotic expression vector pETF1 and pETF2,respectively.The recombinant vectors were then transformed intoE.coliBL21(DE3)for expression under induction of IPTG.SDS-PAGE and Westernblotting showed that F1 and F2 recombinant proteins were successfully expressed and of better reactiongenicity recombinant protein F1 and F2 were successfully purified by the kit of Ni-NTA respectively.The experiment laid a foundation for studying the specific diagnosis kit to detect PPRV infection.
Peste des petits ruminantsvirus;F gene;sequence analysis;prokaryotic expression
S858.2 < class="emphasis_bold">文献标识码:A
A
2010-07-21
家畜疫病病原生物学国家重点实验室开放课题(KEYLAB200802)
倪伟(1982-),女,博士研究生,专业方向为病原分子生物学。
乔军(1971-),副教授,博士,从事分子病毒学研究;e-mail:qj710625@yahoo.com.cn。
猜你喜欢
环球时报(2022-09-20)2022-09-20
今日农业(2020年24期)2020-12-15
天津医科大学学报(2019年6期)2019-08-13
河南畜牧兽医(2017年20期)2018-01-19
现代检验医学杂志(2016年2期)2016-11-14
兽医导刊(2016年12期)2016-05-17
河南畜牧兽医(2015年13期)2015-08-15
现代检验医学杂志(2015年4期)2015-02-06
当代畜禽养殖业(2014年6期)2014-02-27
当代畜禽养殖业(2014年6期)2014-02-27
