
米醋沉淀中Bacillus subtilis DNA提取及RIC-RCR体系条件优化
2011-10-09 02:47廖永红任文雅孙宝国金志刚
食品工业科技 2011年6期
廖永红,任文雅,孙宝国,徐 瑾,沈 晗,金志刚
(北京工商大学,化学与环境工程学院,北京100037)
米醋沉淀中Bacillus subtilis DNA提取及RIC-RCR体系条件优化
廖永红,任文雅,孙宝国*,徐 瑾,沈 晗,金志刚
(北京工商大学,化学与环境工程学院,北京100037)
利用稀释平板法从米醋沉淀中分离获得细菌,通过对其进行16S rDNA分子鉴定,表明该细菌为枯草芽孢杆菌(Bacillus subtilis)。以该菌为实验材料,研究了细菌基因组DNA的提取方法,并将ERIC-PCR法首次应用于醋液菌种,对此反应体系的主要因素进行了优化,最终建立了适合于本实验室的ERIC-PCR体系。结果表明,采用改良的传统细菌基因组DNA提取方法,所提取的DNA质量较高,能够满足ERIC-PCR反应的需要;建立的反应体系为:25μL反应体积10×扩增缓冲液(含 Mg2+)2.5μL,20pmol/μL ERIC-PCR 引物 E1 1.0μL,20pmol/μL ERIC-PCR 引物 E2 1.0μL,DNA模板 2μL,2.5mmol/L dNTPs混合液 1.6μL,taq聚合酶 0.9μL,双蒸水补齐;PCR 反应程序为:94℃ 变性4min,1 个循环;94℃变性 30s,58℃退火 40s,72℃延伸 1min,30 个循环;72℃延伸 4min。
细菌,分离,DNA 提取,ERIC-PCR,优化
食醋在生产过程中需要添加大曲、麦曲、酶母菌及醋酸菌等,各种带菌的原料和工具因操作问题会带来细菌的污染。虽然产品加热灭菌时,一些不耐热的微生物菌体残留在食醋中造成混浊沉淀,一些耐热的芽孢杆菌潜伏在醋中[1],在适当的温度和营养条件下会缓慢生长,影响着米醋的风味及产品的质量。枯草芽孢杆菌(Bacillus subtilis)是一种嗜温性的好氧产芽孢的革兰氏阳性杆状细菌,能够产生耐热、耐旱、抗紫外线和有机溶剂的内生孢子。且枯草芽孢杆菌具有生长快、营养简单以及产生耐热、抗逆芽孢等突出特征。ERIC-PCR(Enterobacterial Repetitive Intergenic Consensus,肠细菌基因间共有重复序列)技术是在随机引物PCR(RAPD)的基础上发展起来的[2-4]。ERIC-PCR 的原理[5-6]是:许多革兰阴性杆菌中存在一段长度为126bp,以多拷贝形式存在的反向重复序列。这段序列分布于基因组中,具有高度的保守性,在不同种间仅有拷贝数和位置变化。依据此重复序列设计引物,提取细菌基因组的总DNA,并对其进行PCR扩增,其保守、独特的位置使这种PCR能产生多种独特的扩增产物(50~3000bp)。扩增产物经琼脂糖凝胶电泳分离形成DNA指纹图谱(DNA迁移带),可根据这些电泳条带来区分细菌的株(型)。在一定范围内,DNA迁移带的大小和多少代表着此重复序列间的距离和重复次数。因此,基因组DNA的差异可清晰显示在指纹图谱上。这样,对混合微生物群落结构的分析就转化为对其DNA指纹图谱的分析。本实验以某企业提供的米醋为材料,进行分离纯化,获得纯菌株进行基因组DNA提取方法的研究和ERIC-PCR反应体系的优化。
1 材料与方法
1.1 材料与仪器
米醋 企业提供的瓶装米醋,肉眼观察有明显沉淀;培养基 土豆、麦芽汁、细菌、营养琼脂培养基、LB 液体培养基[7-8]。
T6型新世纪紫外-可见分光光度计,DK-S22型数显恒温水浴锅,130s超净工作台,LRH-250型生化培养箱,Anymicro Dss显微镜,安莱Alphalmager Hp紫外凝胶成像系统,Dyc-33A型电泳槽,TC-XP-G型PCR热循环仪。
1.2 细菌的分离与纯化
无菌条件下,将米醋瓶盖打开,混合均匀后,定量采取实验醋样,离心收集沉淀。用生理盐水将沉淀打散,选取适宜的稀释度后,以涂布平板法分别涂于土豆、麦芽汁、细菌和营养琼脂培养基平板上,37℃培养48h,检查菌落出现情况。取单菌落,用接种针挖掘一小块菌体移植于另一平板培养基上,37℃培养,再从长有单菌落的平板中选取典型的细菌菌落,取菌体制成悬液,涂布培养、用革兰氏染色法对细菌染色,在显微镜下观察其形态、颜色,并照相,重复培养2~3次以分离纯化菌种。将纯化了的细菌转接到营养琼脂斜面上培养,待生长完好后置于4℃冰箱中保存。
1.3 细菌基因组DNA的提取
1.3.1 3种提取基因组DNA的方法比较 方法一:取单菌落接种于5mL相应的培养基中,在合适的温度下培养。根据细菌的生长率不同,培养时间可由几小时到几天不等。
a.取1.0mL菌液置于1.5mL离心管中,13000r/min离心2min,弃上清。沉淀重悬于1.0mL 0.85%NaCl中,用枪头吸打菌体使之混匀,散开。室温13000r/min离心2min,弃上清。沉淀重悬于550μL 1×TE中。
b.加8μL 蛋白酶 K(20mg/mL),颠倒混匀,37℃温育30min。加40μL 10%SDS,颠倒混匀,37℃温育30min,应为澄清的。加100μL 5mol/L NaCl充分混匀。加 80μLCTAB/NaCl溶 液,混 匀,65℃ 水浴10min。
c.加等体积(0.7~0.8mL)氯仿/异戊醇(24∶1),轻轻振荡混匀。室温,14000r/min离心12min(可看到分三层)。将上清液转移到一支新的1.5mL离心管中,加入等体积酚/氯仿/异戊醇(25∶24∶1)轻轻振荡混匀。14000r/min离心10min。
d.将上清液转移到一支新的1.5mL离心管中,加入等体积氯仿/异戊醇(24∶1)轻轻振荡混匀。14000r/min离心10min。将上清液转移到一新的1.5mL离心管中,加入2倍体积预冷的无水乙醇,颠倒混匀后,-20℃静置30min。室温14000r/min离心8~10min。弃上清,加500μL70%乙醇,轻轻颠倒数次(洗盐)。
e.室温14000r/min离心3min。弃上清,倒置离心管,干燥 DNA沉淀10~15min。DNA沉淀溶于60μL ddH2O 中,-20℃保存备用。
方法二:在方法一的基础上改进而成,即在步骤b和 c之间增加下述步骤,加入8μL 10mg/mL的Rnase,于37℃温浴1h。其余步骤与方法一相同。
方法三:a.取1.0mL菌液于 1.5mL离心管中,13000r/min离心2min,弃上清。沉淀重悬于1.0mL 0.85%NaCl中,用枪头吸打菌体使之混匀,散开。室温13000r/min离心 2min,弃上清。沉淀重悬于550μL 1×TE 中。b.加20μL溶菌酶(100mg/mL),颠倒混匀,37℃温育 30min。c.加 40μL 10%SDS和8μL 10mg/mL 的 Rnase,颠倒混匀,37℃ 温浴 1h。d.细胞匀浆再用8μL蛋白酶K(20mg/mL)消化,37℃温育30min。其余步骤与方法一相同。
1.3.2 DNA质量检测 a.琼脂糖凝胶电泳检测:将所得DNA样品进行0.8%琼脂糖凝胶电泳,采用100V电压,电泳1h,EB染色后,用紫外凝胶成像系统拍照。b.DNA纯度、浓度及提取率检测:取少量DNA样品稀释100倍,用紫外分光光度计测定波长在260、280nm处的吸光光度值。DNA纯度的定性检测,通过A260/A280的比值可以检测所提DNA的纯度。通常纯DNA样品 A260/A280≈1.8。若 A260/A280>1.9,可能有RNA污染;若A260/A280<1.6,可能有蛋白质污染。DNA浓度的计算:
浓度(μg·mL-1)=A260×50 ×稀释倍数
注:1OD 值相当于50μg·mL-1双螺旋 DNA。
1.4 ERIC-PCR反应体系的建立和优化
选取菌种的基因组DNA和ERIC-PCR的通用引物后进行PCR反应体系的建立和优化实验,分别从对TaqDNA聚合酶、dNTPs、引物、退火温度、循环数几个方面做单因素实验,找出各自适合的条件,而且每个合适的条件确定以后都被作为后续研究的一个条件。通过各个因子的组合对ERIC-PCR反应体系的条件进行筛选优化。各个因素的梯度设置见表1。

表1 影响ERIC-PCR反应因素的优化梯度设置
PCR结束后点样于1%的琼脂糖凝胶中电泳,根据条带的有无以及清晰程度和特异性来确定影响因素的最终浓度。ERIC-PCR的程序如下:94℃ 3min;94℃ 30s,58℃ 40s,72℃ 1min,30 个循环;72℃4min;4℃保存。
1.5 16S rDNA分子鉴定
利用1.2的方法将分离得到的菌种进行16S rDNA分子生物学鉴定。
a.利用LB培养基培养3mL新鲜细菌菌液,提取细菌DNA。
b.细菌16S rDNA序列扩增:利用细菌16S rDNA扩增通用引物27F和1492R引物扩增细菌16S rDNA序列。引物信息:27F:5′-GTGCTGCAGAGAGTTTGATCCTGGCTCAG-3′
149 2R:5′- CACGGATCCTACGGGTACCTTGTTACGACTT-3′
c.细菌16S rDNA序列测定:利用引物27F和1492R对细菌16S rDNA序列进行双向测序。测序完成后,对其序列进行拼接。
d.细菌16S rDNA序列比对:登陆NCBI网站,利用Blast软件对细菌16S rDNA序列进行序列比对,获得其最相似细菌,确定样品细菌的种属信息,部分测序工作由北京基诺莱普生物技术有限公司完成。
2 结果与分析
2.1 菌种的分离与纯化
将灭菌后的醋液和未灭菌的醋液分别取0.1mL涂布于土豆、麦芽汁、细菌、营养琼脂培养基上进行好氧和厌氧培养,结果发现,菌种在有氧条件下生长状态优于无氧情况,从营养琼脂和土豆培养基上分离到所要细菌,经过反复纯化后的菌种用于基因组DNA的提取。
2.2 菌种的形态特征
枯草杆菌是芽孢杆菌属的一种。无荚膜,全身鞭毛,能活动。椭圆到柱状,位于菌体中央或稍偏,芽孢形成后菌体不膨大。菌落表面粗糙不透明,污白色或微黄色、在液体培养基中生长时,常形成皱醭,需氧菌。菌种的显微镜照片见图1。
图1 100倍2#菌种显微镜图
2.3 DNA提取方法的比较结果
基因组DNA的提取是进行ERIC-PCR反应的第一步,是进行ERIC-PCR反应的基础条件,所提取的基因组DNA的质量直接关系着ERIC-PCR扩增能否产生清晰和重复性较好的条带。因此,在进行PCR之前有必要对菌种DNA提取方法进行优化。本实验比较了3种细菌基因组提取方法。用紫外分光光度计检测,结果见表2。从DNA提取结果来看,方法一其A260/A280值为1.77。而方法二是在方法一的基础上进行了改进,即多加了Rnase,去除了RNA的干扰,所提取的DNA的质量较高,其A260/A280值为1.81,而且其浓度大小比较适中,能够满足ERIC-PCR的DNA模板的需要。

表2 不同提取方法的DNA浓度及吸光度比值
方法三为溶菌酶方法,溶菌酶可以作用于细菌的细胞壁,具有降解其肽聚糖的作用。从结果上可以看出,方法三A260/A280值为1.46,说明有蛋白的存在,另外此方法所用的时间比较长,而且实验成本也较高,从实际应用的角度上来说并不是最佳的DNA提取方法。所以,综合考虑,方法二为少量提取细菌DNA的最佳方法。
采用方法二提取的基因组DNA经过0.8%的琼脂糖电泳检测,结果如图2所示。

图2 菌种总DNA的凝胶电泳图谱
由图2可以看出,此方法所提取的DNA质量较好而且没有RNA杂带,同时也没有蛋白质和糖类的污染。此基因组DNA可以用于接下来的ERIC-PCR实验。
2.4 ERIC-PCR反应体系影响因素的优化
ERIC-PCR技术的扩增结果受到很多因素的影响。一般能影响PCR扩增的因素均可影响到ERIC-PCR的扩增结果。如Taq聚合酶、dNTPs、引物、退火温度以及循环次数等。因此本实验对以上五个因素进行了单因素优化,建立起适合本实验室的ERIC-PCR体系。
2.4.1 Taq酶的优化 Taq酶的活性与用量直接关系到PCR扩增的成败,是ERIC-PCR反应体系的前提因素。酶量过多易发生非特异反应,而且可能增加突变的机率,尤其在进行高保真扩增时,应尽量减少酶量,但酶量过少时反应性能会下降。Taq酶的浓度如果太低,则合成的产物量减少;如果浓度过高,可能引起非特异性扩增。Taq酶选择的量为0.3、0.5、0.7、0.9、1.1μL,其扩增结果电泳如图 3 所示,由图 3可以看出,Taq酶的量为0.9μL时没有非特异性条带,特征条带在1000bp以下。
2.4.2 三磷酸脱氧核苷酸的影响 dNTP浓度的变化对于ERIC-PCR条带的数量和强弱影响明显,当dNTP浓度不足时,扩增产物减少;而当dNTP浓度过高时,会使扩增错误配对的几率大大增加,过高的dNTP可与Mg2+结合,使得反应体系中Mg2+总量下降,Taq活性受到影响。dNTPs加入量为1.6、1.8、2.0、2.2、2.4μL,其扩增电泳如图 3 所示,可以看出dNTP的量为1.6μL时比较好。2.4.3 引物添加量对ERIC-PCR的影响 引物浓度主要影响扩增产物的特异性,引物浓度变化实质上是改变引物与模板之间的配比几率,从而影响扩增效率。引物浓度如果偏低,就会受到竞争的抑制,引物和模板结合的几率下降,有可能扩增不出来,或者造成条带过浅、缺失等现象。引物浓度过高会引起错误和非特异性产物增加,同时增加引物之间形成二聚体的几率,也可能造成扩增产物的缺失。
本实验采用通用引物进行扩增,梯度设置如下:0.6、1、1.4、1.8、2.2μL,扩增产物电泳结果如图 3 所示,由图3可以看出引物的量为1μL时比较好。

图3 ERIC-PCR条件优化电泳图
2.4.4 退火温度对ERIC-PCR的影响 退火温度是影响PCR反应的重要因素之一,决定着PCR产物扩增的特异性,温度高特异性强,但温度过高则引物不能与模板牢固结合,DNA扩增效率下降;温度低产量高,但过低可造成引物与模板错配,非特异性产物增加,合适的退火温度一般在45~68℃之间。实验设置的温度梯度为:55、56.2、58.0、60.6、63℃,扩增产物电泳结果见图4,由图可以看出,退火温度升高,会减少杂带的产生,但是过高的温度又会造成条带的缺失,所以经过反复的实验后得出最佳退火温度为58℃。
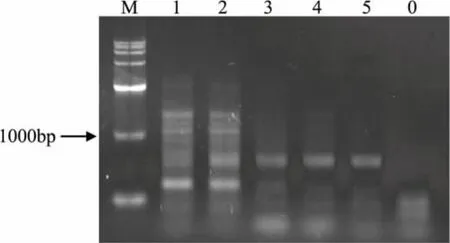
图4 ERIC-PCR退火温度优化电泳图
2.4.5 循环次数对ERIC-PCR的影响 循环次数的设定可根据模板DNA的量、扩增片段的大小和扩增产物的下步应用等因素,设定30~40个循环。循环次数太少,扩增量不足,如果循环次数太多,错配几率会增加,非特异性背景严重。所以,在保证产物得率的前提下,应尽量减少循环次数。本实验设计的循环次数为20、25、30个循环,经实验确定30个循环为最佳循环数。
2.5 ERIC-PCR反应体系的建立
经过对影响ERIC-PCR反应的各个因素的优化,建立了适合于本实验室的 PCR反应体系:在25μL的反应体积中 ERIC-PCR反应体系成分为:25μL反应体积 10×扩增缓冲液(含 Mg2+)2.5μL,20pmol/μL ERIC-PCR 引物 E1 1.0μL,20pmol/μL ERIC-PCR 引物 E2 1.0μL,DNA 模板 2μL,2.5mmol/L dNTPs混合液 1.6μL,taq聚合酶 0.9μL,双蒸水补齐;ERIC-PCR反应程序为:94℃变性4min,1个循环;94℃变性 30s,58℃退火 40s,72℃延伸 1min,30 个循环;72℃延伸4min。
2.6 16S rDNA分子鉴定结果
登陆 NCBI网站,利用 Blast软件对细菌16S rDNA序列进行序列比对,获得其最相似细菌,确定样品细菌为枯草芽孢杆菌。NCBI登录号为AL009126,相似度为96%。
3 讨论
基因组DNA的质量对ERIC-PCR反应有着重要的影响,它决定了PCR的成功与否。而提取的DNA的方法因实验材料的不同也有所区别,本实验比较了3种提取DNA的方法,基本上是传统方法和溶菌酶方法的比较,溶菌酶的方法提取的DNA的总量很高但是纯度不高。方法二是在传统方法上进行了改进,在中间的过程中加入了Rnase降解步骤,基本上解决了RNA污染的问题,是提取蜡状芽孢杆菌的最适合方法。方法三加入了Rnase和溶菌酶,但是由于成本比较高和时间相对长而受到限制。
ERIC-PCR技术具有DNA样品需要量少,引物价格便宜,成本较低、操作技术简单等特点。但由于其使用的是通用引物,所以合适的PCR条件是决定成功与否的关键性因素。扩增条件的变化会对ERIC-PCR图谱产生很大的影响,从而影响ERIC-PCR分析的准确性。而条带的准确性是进行PCR分析的重要前提条件。在PCR反应中由于反应体系不适宜则会出现两种极端的现象,一种是非特异性扩增严重,产生许多不确定的产物甚至无目的条带,另一种是扩增不到任何产物。这时需要通过改变Taq酶的用量、dNTP浓度、引物用量、Mg2+浓度、退火温度、循环次数等加以优化。本实验采用了单因素递进筛选法,以上一轮筛选出的值用于下一个因素的筛选,层层递进的方式,经过比较和分析,建立稳定且适合该菌种的ERIC-PCR体系为:25μL反应体积10×扩增缓冲液(含 Mg2+)2.5μL,20pmol/μL ERIC-PCR 引物E1 1.0μL,20pmol/μL ERIC- PCR 引物 E2 1.0μL,DNA 模板 2μL,2.5mmol/L dNTPs混合液 1.6μL,taq聚合酶0.9μL,双蒸水补齐;ERIC-PCR反应程序为:94℃变性 4min,1 个循环;94℃ 变性 30s,58℃ 退火40s,72℃延伸 1min,30 个循环;72℃延伸4min。
[1]王先秀.食醋细菌性浑浊问题的探讨[J].中国酿造,1997(1):6-8.
[2]Sharples GJ.Nucleic Acids Res[J],1990,18:6503-6508.
[3]Hulton CS.Mol Microbiol[J],1991(5):825-834.
[4]Versalovic J,Koeuth T.Distribution ofreptitive DNA sequences in eubacteria and application to fingerprinting of bacteria genomes[J].Nucleic Acids Res,1991,19(24):6823-6831.
[5]Giovanni D I,Lidia S,Watrud RJ,et al.Fingerprinting of mixed bacterial strains and BOILOG gramnegative(GN)substrate communities bentero bacterial repetitive intergenic consensus sequence-PCR(ERIC-PCR)[J].Cur Microbiol,1999,38:217-223.
[6]Gillings M,Holley M.Repetitive element PCR fingerprinting(REP-PCR)using enterobacterial repetitive intergenic consensus(ERIC)primers is not necessarily directed at ERIC elements[J].Lett Appl Microbiol,1997,25(1):17-21.
[7]杜连祥.工业微生物学实验技术[M].天津:天津科学技术出版社,1992:236-237.
[8]陈天寿.微生物培养基的制造与应用[M].北京:中国农业出版社,1995:67-70.
[9]王先秀.食醋细菌性浑浊问题的探讨[J].中国酿造,1997(1):6-8.
[10]陆正清,王艳.食醋的混浊及其防治[J].江苏调味副食品,2003(4):14-17.
[11]张宗舟,张海林,陈志梅.发酵工艺对食醋浑浊影响的研究[J].中国酿造,2006(1):12-13.
[12]贾莉.应用酶技术解决山西老陈醋浑浊沉淀的研究[D].山西:山西农业大学硕士论文,2003:9-10.
Genomic DNA extraction from Bacillus subtilis in vinegar precipitation and the optimization of the system of ERIC-PCR
LIAO Yong-hong,REN Wen-ya,SUN Bao-guo*,XU Jin,SHEN Han,JIN Zhi-gang
(College of Chemical and Environmental Engineering,Beijing Technology and Business University,Beijing 100037,China)
Bacillus subtilis were isolated by streak platemethod.It was identified that was Bacillus subtilis by using the 16S rDNA gene.Bacillus strains were used as experimental materials.The method of genomic DNA extraction was studied in Bacillus and the conditions of ERIC-PCR.Finally,ERIC-PCR reaction system was suitable for our lab was established.The results showed that high-grade genomic DNA which could meet the requirements of PCR reaction was obtained by the modified methods.The established ERIC-PCR reaction system was as follows:10×Buffer(Mg2+)2.5μL,20pmol/μL primer E1 1μL,20pmol/μL primer E2 1μL,DNA template 2μL,2.5mmol/L dNTPs 1.6μL,Taq polymerase 0.9μL,ddH2O 16μL,25μL reaction volume.The reaction program of PCR was devised as follows:4min degeneration at 94℃(one cycle),then 30s degeneration at 94℃,40s annealing at 58℃,and 1min extension at 72℃(30 cycles),and then 4min final extension at 72℃.
bacillus;isolation;DNA extraction;enterobacterial repetitive intergenic consensus-PCR(ERICPCR);optimization
TS201.3
A
1002-0306(2011)06-0212-05
2010-05-06 *通讯联系人
廖永红(1965-),女,副教授,主要从事有关食品发酵工程方面的教学和科研工作。
国家“十一五”科技支撑项目。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
军事文摘(2021年18期)2021-12-02
今日农业(2021年11期)2021-08-13
罕少疾病杂志(2017年2期)2017-02-23
广西林业科学(2016年3期)2016-03-16
实用手外科杂志(2015年3期)2015-08-27
山西农经(2015年7期)2015-07-10
食品工业科技(2014年15期)2014-03-11
遗传(2014年3期)2014-02-28
