
冠心生脉口服液质量标准研究
2011-09-17 06:40李海燕
中国药业 2011年1期
李海燕,康 建
(1.河南省食品药品检验所,河南 郑州 450003; 2.郑州大学第一附属医院,河南 郑州 450052)
冠心生脉口服液被收载于《卫生部药品标准新药转正标准(第十册)》,具有益气生津、活血通脉的功效,用于治疗心气不足、心阴虚引起的心悸气短、胸闷作痛、自汗乏力、脉微结代。全方由人参、麦冬、五味子(醋炙)、丹参、赤芍、郁金、三七等药材组成。原标准中的质量控制方法较简单且操作烦琐,不能有效控制药品质量。笔者根据处方所含药味的化学成分及剂型特点,研究制订了赤芍、麦冬、人参及三七、五味子的薄层色谱鉴别及赤芍中芍药苷的含量测定[1]方法。现报道如下。
1 仪器与试药
Waters 2690型高效液相色谱仪;Waters 996型二极管阵列检测器;硅胶G预制薄层板(青岛海洋化工厂,批号为030507);超声波清洗机(功率250 W,频率40 kHz)。麦冬、人参、三七对照药材(批号分别为 121013-200607,120917-200609,120941-200506)及芍药苷对照品(批号为 110736-200731,供含量测定用,以97.9%计算)、五味子醇甲对照品(批号110857-200608)均由中国药品生物制品检定所提供;冠心生脉口服液(批号分别为081001,081002,081003,河南省宛西制药股份有限公司);甲醇、乙腈为色谱纯,水为乐百氏纯净水,其他试剂均为分析纯。
2 方法与结果
2.1 薄层色谱鉴别
赤芍:取本品10 mL,置分液漏斗中,用正丁醇10 mL振摇提取,将正丁醇液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液;按冠心生脉口服液制备工艺制备不含赤芍的阴性对照品溶液;另取芍药苷对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。照薄层色谱法试验,吸取上述3种溶液各10 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(40 ∶5 ∶10 ∶0.2)为展开剂,展开,取出,晾干,喷以 5% 香草醛硫酸溶液,加热至斑点显色清晰。结果供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液无干扰(图 1 A)。
麦冬:取本品10 mL,加盐酸溶液(18→100)1 mL,置水浴上浓缩至约5 mL,用三氯甲烷10 mL振摇提取,将三氯甲烷液浓缩至2 mL,作为供试品溶液;按冠心生脉口服液制备工艺制备不含麦冬的阴性对照品溶液;另取麦冬对照药材1 g,加水30 mL,加热回流30 min,滤过,滤液浓缩至10 mL,同法制成对照药材溶液。照薄层色谱法试验,吸取上述3种溶液各10 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-丙酮(4∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,加热至斑点显色清晰。结果供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液无干扰(图1 B)。
人参及三七:取本品30 mL,用乙酸乙酯30 mL振摇提取,水液用水饱和的正丁醇提取振摇2次,每次30 mL,合并正丁醇液,用氨试液洗涤2次,每次30 mL,正丁醇液用正丁醇饱和的水30 mL洗涤,正丁醇液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液;按冠心生脉口服液制备工艺制备不含人参和三七的阴性对照品溶液;另取人参、三七对照药材各1 g,分别加甲醇25 mL,加热回流1 h,放冷,滤过,滤液蒸干,残渣加水20 mL使溶解,分别同法制成对照药材溶液。照薄层色谱法试验,吸取供试品溶液5 μL、人参对照药材溶液及三七对照药材溶液各1 μL,分别点于同一硅胶G薄层板上,以10℃下放置的三氯甲烷-甲醇-水(13∶7∶2)的下层溶液为展开剂,在2~10℃展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。结果供试品溶液色谱中,在与对照药材溶液色谱相应位置上分别显相同颜色的斑点,阴性对照品溶液无干扰(图1 C)。
五味子:取本品50 mL,用三氯甲烷振摇提取2次,每次30 mL,将三氯甲烷液蒸干,残渣加三氯甲烷1 mL使溶解,作为供试品溶液;按冠心生脉口服液制备工艺制备不含五味子的阴性对照品溶液;另取五味子醇甲对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。照薄层色谱法试验,吸取供试品溶液10 μL,对照品溶液1 μL,分别点于同一硅胶GF254薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(10∶5∶1)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(254 nm)下检视。结果供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照品溶液无干扰(图1 D)。

图1 薄层色谱图
2.2 含量测定
2.2.1 色谱条件与系统适用性试验
色谱柱:Agilent Eclipse XDBC18柱 (250 mm ×4.6 mm,5 μm);柱温:35℃;检测波长:230 nm;流动相:乙腈-水(14∶86)[2];流速:1 mL/min;进样量:10 μL。理论板数按芍药苷峰计算应不低于5 000。在此色谱条件下的色谱图见图2。
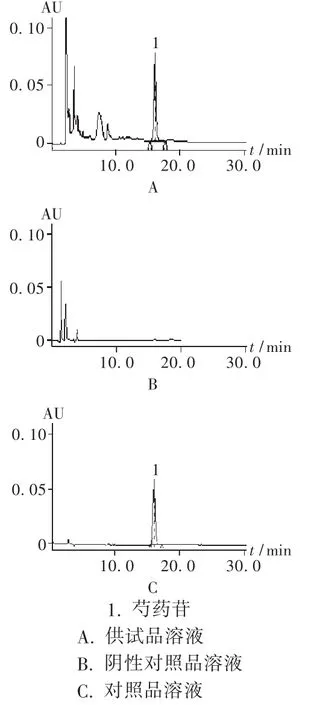
图2 高效液相色谱图
2.2.2 溶液制备
精密称取芍药苷对照品适量,精密称定,加甲醇制成每1 mL含0.1 mg的对照品溶液;精密量取本品 10 mL,置 100 mL量瓶中,加30%甲醇稀释至刻度,摇匀,作为供试品溶液;按冠心生脉口服液制备工艺和供试品溶液制备方法制备不含赤芍的阴性对照品溶液。
2.2.3 方法学考察
线性关系考察:精密吸取芍药苷对照品溶液(406.5 μg/mL)1,3,5,7,10,13 μL,按上述色谱条件测定。以进样量(X)为横坐标、峰面积积分值(Y)为纵坐标绘制标准曲线,得回归方程 Y=1 445 284X-47 609,r=0.999 3(n=6)。结果表明,芍药苷进样量在 0.406 5 ~5.284 5 μg范围内与峰面积线性关系良好。
精密度试验:精密吸取同一供试品溶液10 μL,重复进样6次,测定峰面积。结果的 RSD为0.09%(n=6)。
重复性试验:对同一批(批号为081001)样品5份进行测定。结果的 RSD为0.33%(n=5),表明方法重复性良好。
稳定性试验:取同一供试品溶液,按上述色谱条件分别于0,2,4,6,8,10 h时测定芍药苷峰峰面积积分值。结果的 RSD为0.34%(n=6),表明供试品溶液在10 h内基本稳定。
加样回收试验:取已知含量的同一批(批号为081001)样品6份各5 mL,每份加对照品贮备液适量,按供试品溶液制备方法制备加样供试品溶液,进样测定,计算加样回收率。结果见表1。

表1 芍药苷加样回收试验结果(n=6)
2.2.4 样品含量测定
分别取批号为 081001,081002,081003的 3批样品,按“2.2.2”项下方法制成供试品溶液,进样测定,按外标法计算含量。结果 3 批样品中芍药苷含量分别为 1.126,1.135,1.126 g/L。
3 讨论
本品既含人参又含三七,用普通硅胶G薄层板无法对二者进行鉴别。笔者曾尝试鉴别三七,以三七皂苷R1和三七对照药材为对照,使用高效硅胶G薄层板(青岛海洋化工厂),用10℃以下放置的三氯甲烷-甲醇-水(13∶7∶2)的下层溶液为展开剂,在2~10℃展开,还做了二次展开试验,但分离效果和重现性不佳。因此,以人参、三七对照药材同时作为对照,用普通硅胶G薄层板做鉴别试验;试验中曾以五味子对照药材为对照,结果供试品溶液色谱中在与缺五味子的阴性对照的相应位置上有相同颜色的主斑点出现,阴性对照有干扰。
含量测定选用溶液直接稀释的方法,操作简易。比较了30%甲醇溶液、50%甲醇溶液、70%甲醇溶液及甲醇的提取效果。用甲醇作溶剂时,样品液浑浊,用30%甲醇溶液、50%甲醇溶液及70%甲醇溶液提取时,样品液澄清,因此选用30%甲醇溶液为提取溶剂。
[1]王亚林.HPLC测定宫炎康颗粒中芍药苷的含量[J].陕西中医学院学报,2006,29(4):41.
[2]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2005:405.
猜你喜欢
食品安全导刊(2021年36期)2021-03-14
科学导报(2020年75期)2020-12-21
中成药(2017年8期)2017-11-22
海峡科技与产业(2016年3期)2016-05-17
中国民族医药杂志(2016年9期)2016-05-09
中国民族医药杂志(2016年4期)2016-05-09
中国当代医药(2015年8期)2015-03-01
中国药业(2014年24期)2014-05-26
中国合理用药探索(2014年11期)2014-03-11
中国中医药现代远程教育(2014年14期)2014-03-01
