
镍基催化剂对CO-超临界水制氢固碳反应的影响
2011-08-15 08:00赵永椿张军营吕涛涛郑楚光
动力工程学报 2011年11期
田 冲, 赵永椿, 张军营, 晏 恒, 吕涛涛, 郑楚光
(华中科技大学煤燃烧国家重点实验室,武汉 430074)
二氧化碳引起的“温室效应”已成为全球关注的热点,如何有效地控制CO2的排放成为当务之急.目前,控制CO2排放的主要措施集中在以下三方面:(1)提高燃料的利用效率;(2)开发低碳或无碳能源;(3)将产生的CO2捕集、吸收并封存起来,减少排入大气中的CO2量[1].在开发无碳能源方向,氢能因其洁净、来源广泛和燃烧后无污染等特点,被广泛认为将成为未来的新能源载体.H2可通过天然气、石油、生物质、太阳能、核能、垃圾和煤炭等多种能源产生.以氢能为基础的未来能源结构被认为是理想的长期应对环境问题的能源结构[2].
目前,大批国内外学者[3-8]致力于含碳能源直接制氢的研究.在超临界状态下,水的性质近似于非极性有机溶剂,可与大多数有机物及气体互溶,形成均相反应环境.与常压气化制氢相比,超临界水制氢在制氢领域占有越来越重要的地位[6].Lin等[3-5]提出将煤和氧化钙混合后在水蒸气条件下气化制氢的研究思路,制定了基于超临界的 HyPr-RING的试验研究和开发计划,实现了制氢过程中同时脱除二氧化碳及循环利用固碳剂.国内外学者[7-9]在生物质/煤-超临界水制氢方面也做了许多研究工作,研究重点主要集中于如何提高制氢效率.大量研究[10-14]表明,镍基催化剂虽然能够促进水气转化反应中制氢效率的提高,但是其作用机理尚未完整建立.
笔者在硅灰石直接碳酸化隔离二氧化碳研究的基础上[15-16],添加水热法合成催化剂 Ni/ZrO2-CeO2-Al2O3,进行了 CO-超临界水气转换反应试验,分析了Ni/ZrO2-CeO2-Al2O3催化剂和硅灰石固碳剂对制氢效率及CO2矿物碳酸化效率的影响,为含碳能源制氢固碳一体化技术提供了试验基础.
1 试验方法
1.1 水热法合成Ni/ZrO2-CeO2-Al2O3
按化学剂量比准确称取一定量的Ce(NO3)3◦6H2O、ZrOCl2◦8H2O、Al(NO3)3◦9H2O 和Ni(NO3)2◦6H2O,在烧杯中混合后用去离子水溶解,滴加氨水调节混合溶液的pH值至2.5,将混合溶液放入体积为80 ml、温度为115℃的高压釜中加压恒温密闭24 h.取出后将其在70℃的水浴中蒸至粘稠状,在115℃下烘干后在800℃的马弗炉中煅烧8 h,即得到褐色的Ni/ZrO2-CeO2-Al2O3粉末.利用 x′pert PRO X射线衍射仪(XRD)和 EAGLE III X-射线荧光能谱仪(XRF)对催化剂组成进行表征.
1.2 CO-超临界水试验
高温高压条件下,由于超临界水的强反应活性,CO与超临界水会发生水气转化(Water-Gas Shift,WGS)反应;在系统中加入硅灰石(主要成分为Ca-SiO3)后,CaSiO3与水气转化反应生成的CO2发生固碳反应,主要反应如下:

CO-超临界水制氢固碳所用ESTANIT-GMBH高压反应釜的参数:pmax=35 MPa,Tmax=500℃,容积为300 mL,系统配备自动测压测温控制仪表,反应气体为CO.试验过程中,按一定的化学剂量精确称取催化剂Ni/ZrO2-CeO2-Al2O3和固碳剂硅灰石置于高压反应釜中,添加100 mL去离子水,密闭反应釜后充入一定量的CO气体排尽反应釜内残存的空气,调节阀门开度后继续通入CO,直到CO的初始压力达到稳定.通入CO一方面是在反应体系中提供反应物,另一方面则是避免水在加热过程中过早汽化为水蒸气而导致加热效果变差.一切就绪之后,启动电炉加热,升温至设定温度并保温 30 min.反应结束后,通过空气自然冷却收集气相产物,采用气相色谱测定其组分;固体产物经过滤、干燥后进行XRD测试和煅烧试验,分析其组分变化情况及确定硅灰石对CO2的吸收效果,具体试验工况如表1所示.在保温过程中,每种工况条件下温度升至设定温度后,压力仪表控制面板上显示的系统压力均在24 MPa以上.达到设定反应温度后,随着CO初压增加,系统压力也随之增加,当CO初压为6 MPa,温度为400℃时,保温反应阶段中系统压力达32.1 MPa.
2 结果与讨论
2.1 固碳剂样品
试验中所用的浙江长兴硅灰石化学成分分析如表2所示,其中 CaSiO3硅酸钙质量分数达到95%[16].矿石经粉碎后研磨,过200目标准筛,650℃空气气氛焙烧2 h之后备用.
2.2 Ni/ZrO2-CeO2-Al2O3的表征
图1给出了催化剂Ni/ZrO2-CeO2-Al2O3的XRD谱图.图1中出现了较强的衍射峰,表明在该条件下各物质相互之间发生了掺和,形成较完善的晶相结构,制备了具有一定活性的催化剂.当衍射角为 29°、33.4°、48.3°和 57.4°时,出现的明显衍射峰为CeO2,35.5°和60°时为ZrO2衍射峰,NiO 的峰较弱,没有检测到明显的Al2O3相.
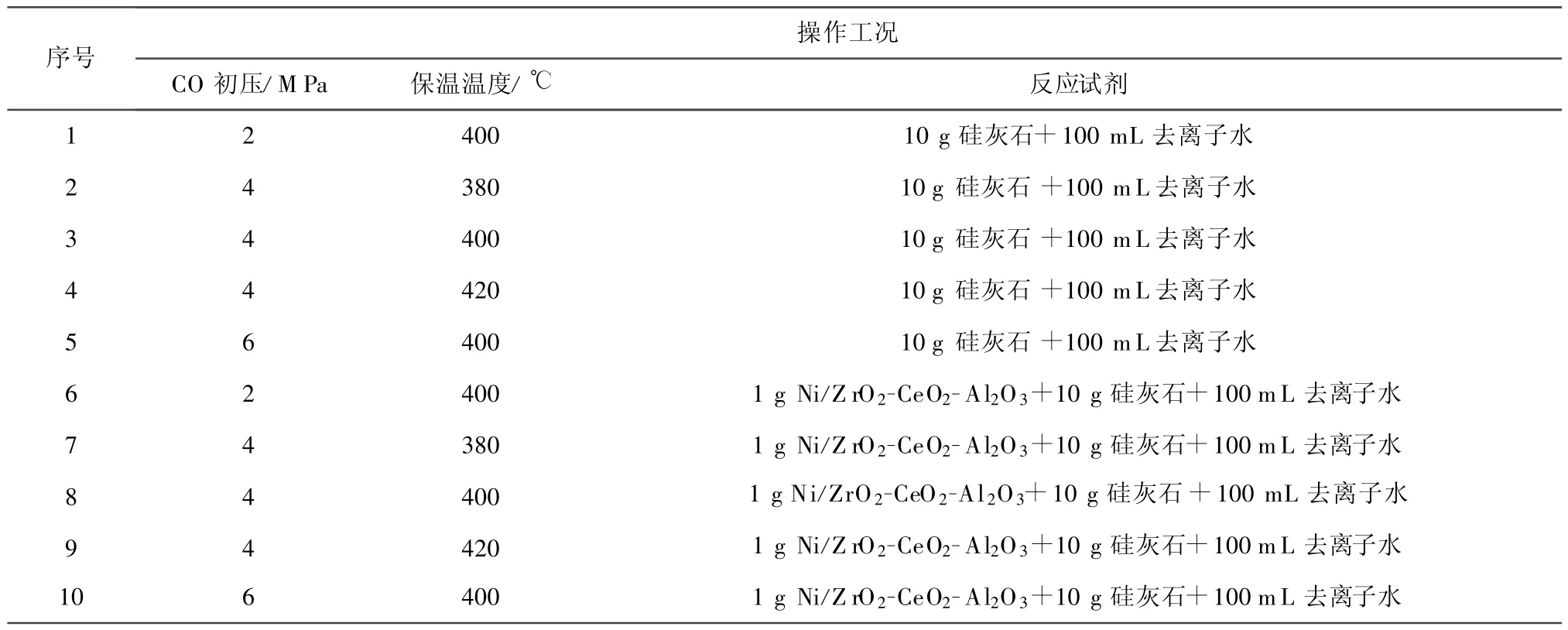
表1 CO-超临界水反应工况Tab.1 CO-supercritical water-gas shift reaction conditions

表2 硅灰石组分Tab.2 Chemical composition of wollastonite
对催化剂Ni/ZrO2-CeO2-Al2O3进行XRF分析,得到催化剂的各组分分析(见表3),其中Ni的质量分数为10%左右。
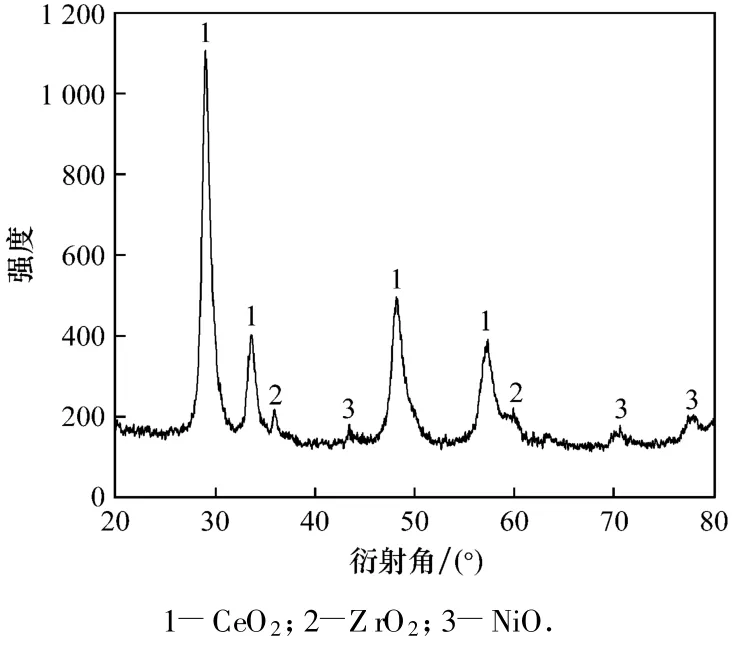
图1 Ni/ZrO2-CeO2-Al2O3的XRD图谱Fig.1 XRD pattern of Ni/ZrO2-CeO2-Al2O3
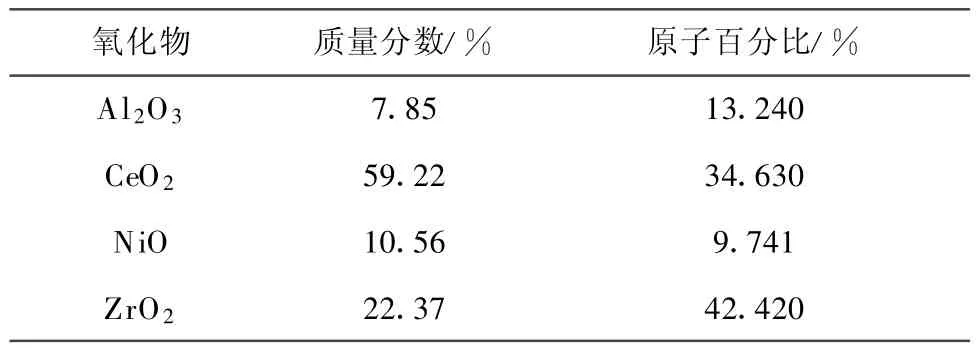
表3 催化剂成分分析Tab.3 Composition analysis of catalyst
2.3 矿物碳酸化固体产物XRD分析

图2 反应后矿物碳酸化固体产物XRD分析Fig.2 XRD spectra of wollastonite carbonation products
图2给出了工况4和工况9两种工况下反应后的矿物碳酸化固体产物XRD图谱.对比反应前后矿石组分可见,碳酸钙和二氧化硅存在于反应后的矿石组分中.这一结果表明:CO-超临界水条件下,试验过程中添加硅灰石对反应产生的CO2进行了碳酸化固定,即发生了反应(2).
2.4 煅烧结果
先将反应的固相产物置于105℃烘箱中干燥48 h,再在马弗炉中对其进行高温热分解试验.因为碳酸钙的热分解温度在800℃左右,设定马弗炉的煅烧温度为600℃和800℃,每个温度段停留1 h,记录固体产物的质量变化.为保证结果的准确性,对同一个样品进行两次高温热分解试验,取其矿化效率的平均值.
二氧化碳硅灰石矿化效率η[16]为:
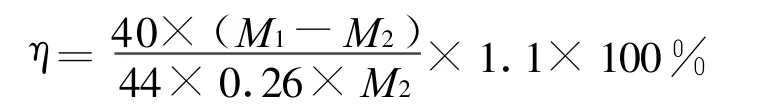
式中:M1为煅烧前所取固体产物的质量,g;M2为煅烧后固体产物的剩余质量,g;1.1为修正值.
此试验结果进一步定量地验证了2.3节是硅灰石对CO2碳酸化固定的效果.表4列出了每种工况下热分解试验后的CO2矿化效率.结果表明:硅灰石能实现CO2的矿物碳酸化固定,且不同操作条件下CO2的矿化效率不同.
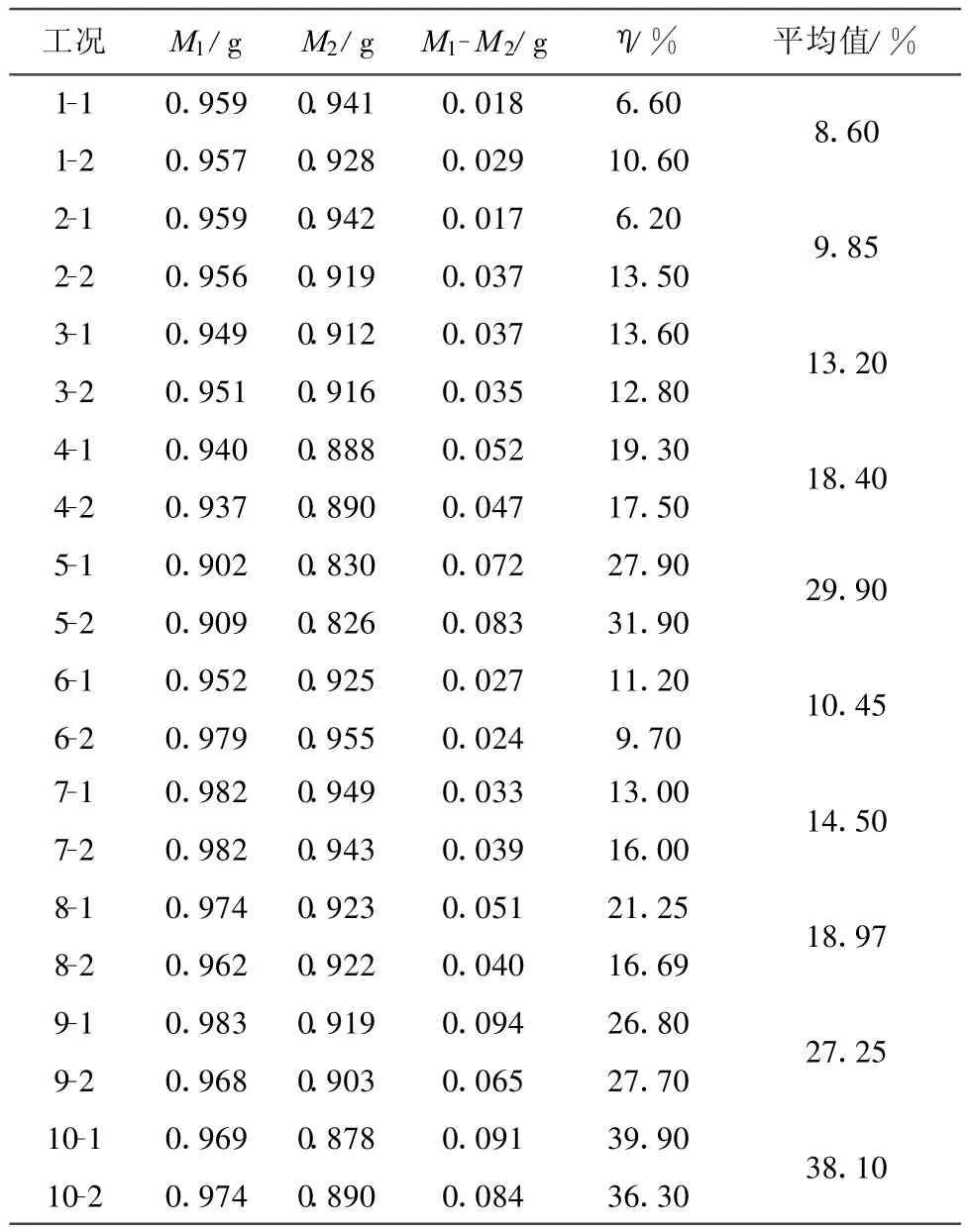
表4 CO2矿化效率Tab.4 CO2mineral carbonation rate
2.5 影响制氢效率的因素分析
2.5.1 温 度
图3给出了CO的初压为4 MPa,反应温度分别为380℃、400℃和420℃时产物中H2的体积分数即制氢效率随温度变化的曲线.从图3可知,温度的升高有利于CO-超临界水WGS反应的进行.这是因为:(1)温度升高加快了水气转化反应速度,也提高了CO和超临界水的反应活性,因而气体产物中H2的体积分数随着温度的升高而增加;(2)反应温度升高,硅灰石矿石活性增加、矿石溶解速度加快,矿化反应速率常数随着温度的升高而增大;(3)CO与超临界水反应生成的CO2体积分数也会随之增加,促进反应(2)的进行,CO2消耗量增加,使水气转换反应平衡向有利于H2生成的方向移动,硅灰石矿化效率升高的同时提高了制氢效率.
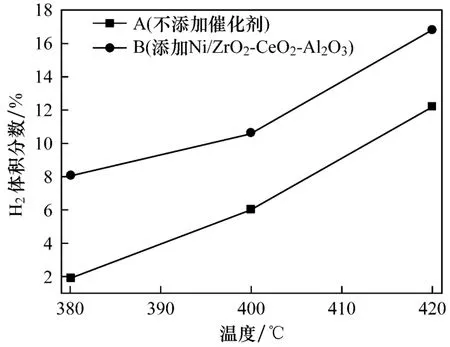
图3 CO初始压力为4 MPa时不同温度下的制氢效率Fig.3 H2production at different temperatures for an initial CO pressure of 4 MPa
催化剂Ni/ZrO2-CeO2-Al2O3的存在对提高制氢效率具有一定的促进作用.每种试验工况下,催化剂都表现出一定的活性,在CO初压为4 MPa、反应温度为420℃时,制氢效率最高达到16.8%,比相同工况下不添加催化剂时提高了4.6%.
2.5.2 CO初压
图4给出了反应温度为400℃,CO初压分别为2 MPa、4 MPa和6 MPa时,反应后制氢效率随压力变化的曲线.从图4中两条曲线的变化规律可知,CO初压增大能够促进CO-超临界水WGS反应的进行,提高反应后气相产物中的H2体积分数.这是因为:(1)随着CO初压的增加,CO-超临界水体系中CO的体积分数升高,有利于WGS反应平衡朝正反应方向进行,促进了H2的生成,提高了制氢效率;(2)CO与超临界水反应生成CO2的体积分数也会相应增大,促进CO2与硅灰石作用生成碳酸盐,消耗CO2的同时使得水气转化反应朝有利于H2生成的方向移动,提高了产物中的H2体积分数.比较图4中两条曲线可知,添加催化剂Ni/ZrO2-CeO2-Al2O3对制氢效率有一定的提高,在CO初压为6 MPa,反应温度为400℃时,制氢效率最高达到14.2%,比相同工况下不添加催化剂条件时提高了8.2%.
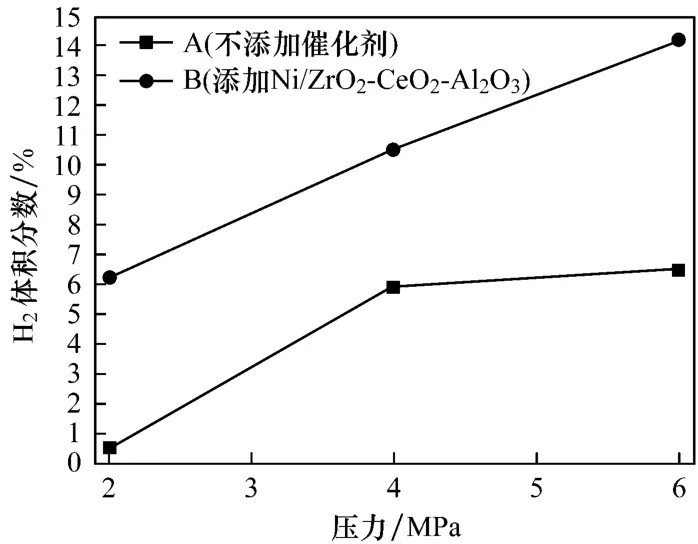
图4 CO温度为400℃时不同压力下的制氢效率Fig.4 H2production at different pressures fo r a temperature of 400℃
2.5.3 不同工况下制氢效率的比较
图5给出了不同工况下制氢效率的对比,图中柱1、2、3、4和 5表示不添加催化剂时反应后气相产物中的H2体积分数,与之对应的柱6、7、8、9和10表示相同工况下添加催化剂Ni/ZrO2-CeO2-Al2O3时反应后气相产物中的H2体积分数.当催化剂Ni/ZrO2-CeO2-Al2O3存在时,所有工况下制氢效率都有一定的提高.试验结果表明:添加催化剂Ni/ZrO2-CeO2-Al2O3使得CO-超临界水WGS反应表现出一定的活性.在CO初压为6 MPa,温度为400℃时,制氢效率增幅最大,达到7.7%.对比同样压力下的试验工况可知,随着温度的增加,制氢效率增幅有一定的变化.由此推测,催化剂活性随着温度的增加呈减弱趋势,但在试验所选的工况条件下,变化并不明显.这是因为温度升高时镍基催化剂容易发生积碳现象,影响催化剂的活性,需要通过调控制备方法来加以改善[14].对比相同温度工况下的试验结果可知:压力对催化剂的活性有一定的影响,压力越大,催化剂活性越高,制氢效率越高.
图5 不同工况下的制氢效率Fig.5 H2production under different working conditions
2.6 制氢固碳一体化
2.6.1 CO2矿化效率对比
图6给出了不同工况下CO2的矿化效率.对比图6中柱A与柱B可知,当Ni/ZrO2-CeO2-Al2O3存在时,CO2的矿化效率有所提高.这是因为CO-超临界水WGS反应后产生了较高浓度的 H2,同时CO2的浓度也随之升高,促进了硅灰石与CO2之间的碳酸化反应,提高了CO2的矿化效率.由上述分析可知,镍基催化剂的存在不但不会抑制硅灰石对CO2的碳酸化作用,反而在一定程度上促进了硅灰石对CO2的碳酸化反应,提高硅灰石的矿化效率.
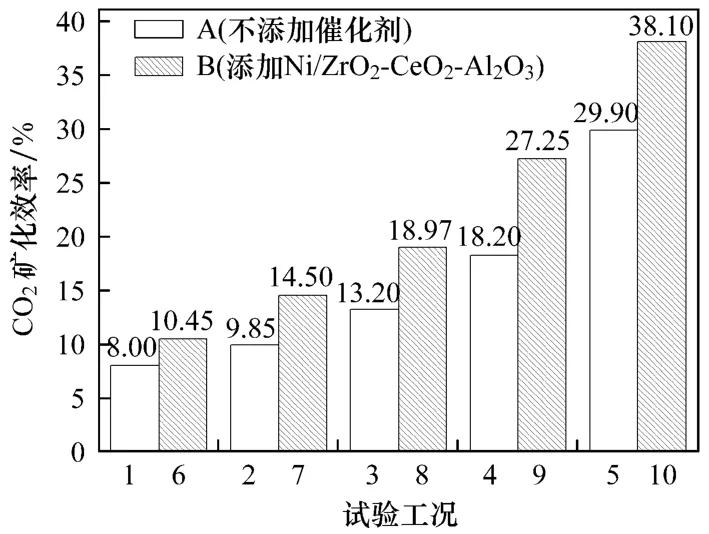
图6 不同工况下CO2的矿化效率Fig.6 CO2sequestration rate under different working conditions
2.6.2 制氢效率与CO2矿化效率变化趋势对比
图7给出了当催化剂 Ni/ZrO2-CeO2-Al2O3存在时,各种工况下的制氢效率和CO2的矿化效率.在工况6、工况7、工况8和工况9这4种情况下,制氢效率与CO2矿化效率的变化基本一致,制氢效率得到了提高,同时CO2矿化效率也随之升高.当CO初压为6 MPa,温度为400 ℃时(即工况10),在制氢效率没有提高的情况下,CO2矿化效率却有了一定幅度的提高.这一现象表明,提高反应温度和增加CO初压都有利于CO-超临界水WGS制氢固碳反应,但两者对制氢和固碳的影响程度不同.
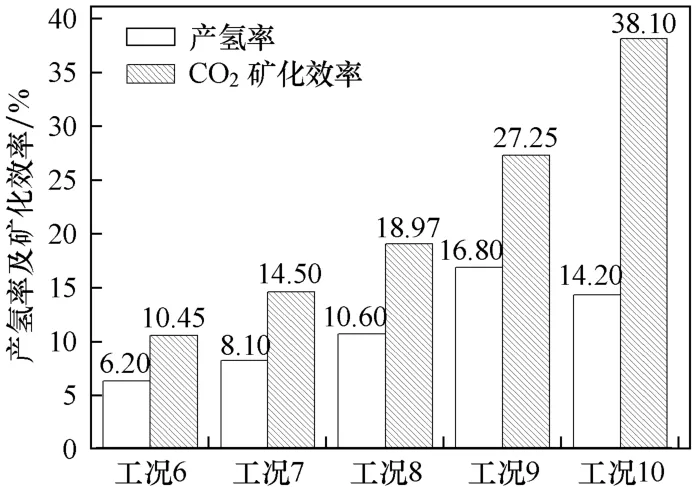
图7 不同工况下的产氢率和CO2矿化效率Fig.7 H2production and CO2sequestration rate under different working conditions
分别将工况9和工况10与工况8进行对比可知:CO初压同为4 MPa时,420℃较之400℃制氢效率提高6.2%,矿化效率提高8.28%;当CO初压为6 MPa时的制氢效率比初压为4 MPa时降低3.6%,矿化效率提高19.13%.结果表明:CO初压对CO2矿化效率影响较大,反应温度对制氢效率的影响较为明显.这是因为CO初压增加,导致保温过程中系统压力增加,这样使得反应体系中CO2分压有所提高,硅灰石在超临界水中的溶解速率增加,促进了硅灰石与CO2的反应,从而提高了硅灰石的矿化固碳效率.为了同时提高制氢效率和CO2矿化效率,实现制氢固碳一体化的试验研究需要综合考虑CO初压和反应温度2个因素,折衷选择最优工况.
3 结 论
(1)同时添加镍基催化剂和硅灰石的条件下发生CO-超临界水WGS反应,生成H2,同时硅灰石对CO2进行碳酸化固定.
(2)水热法合成催化剂 Ni/ZrO2-CeO2-Al2O3对CO-超临界水WGS制氢固碳反应有一定的促进作用,能同时提高产物中的H2体积分数和CO2矿化效率.
(3)当CO初压为4 MPa,温度为420℃时,添加1 g催化剂和10 g硅灰石样品,制氢效率最高达到16.8%;当CO初压为6 MPa,温度为400℃时,添加1 g催化剂和10 g硅灰石样品,CO2矿化效率最高达到38.1%.
(4)提高温度和CO初压对CO-超临界水WGS制氢固碳反应均有促进作用,在存在催化剂Ni/ZrO2-CeO2-Al2O3的条件下,CO初压对CO2矿物碳酸化的影响较大,温度对制氢效率的影响较大.
[1]郑楚光.温室效应及其控制对策[M].北京:中国电力出版社,2001.
[2]NAVA RRO R M,PEÑA M A,FIERRO J L G.Hydrogen production reactions from carbon feedstocks:fossil fuels and biomass[J].Chemical Reviews,2007,107(10):3952-3991.
[3]LIN S Y,SUZUKI Y,HATANO H,et al.Hydrogen production from organic material reaction with supercritical water accompanied by CO2adsorption[J].Kagaku Kogaku Ronbunshu,1999,25(3):498-500.
[4]LIN S Y,SUZUKI Y,HATANO H,et al.Hydrogen production from hydrocarbon by integration of water-carbon reaction and carbon dioxide removal(HyPr-RING method)[J].Energy Fuels,2001,15(2):339-343.
[5]LIN S Y,HARADA M,SUZUKI Y,et al.Hydrogen production from coal by separating carbon dioxide during gasification[J].Fuel,2002,81(16):2079-2085.
[6]银建中,王伟彬,张传杰,等.超临界水处理生物质制氢技术[J].生物技术,2007,17(3):92-95.YIN Jianzhong,WANG Weibin,ZHANG Chuanjie,et al.The hydrogen production methods from biomass using catalysis gasification in supercritical water[J].Biotechnology,2007,17(3):92-95.
[7]LU Y J,GUO L J,JI C M,et al.Hydrogen produc-tion by biomass gasification in supercritical water:a parametric study[J].International Journal of Hydrogen Energy,2006,31(7):822-831.
[8]王树众,王亮,公彦猛,等.煤的超临界水热氧化反应动力学及系统热能的研究[J].动力工程,2009,29(6):565-570.WANG Shuzhong,WANG Liang,GONG Yanmen,et al.Studies on reaction kinetics of coal's thermal oxidation in supercritical water and thermal energy of the System[J].Journal of Power Engineering,2009,29(6):565-570.
[9]CHENG Leming,ZHANG Rong,BI Jicheng.Pyrolysis of a low-rank coal in sub-and supercritical water[J].Fuel Processing Technology,2004,85(8/9/10):921-932.
[10]HARYANTO A,FERNANDO S D,FILIP S D,et al.Hydrogen production through the water-gas shift reaction thermodynamic equilibrium versus experimental results over supported Ni catalysts[J].Energy&Fuels,2009,23(6):3097-3102.
[11]WEN W,CALDERON J E,BRITO J L,et al.In situ time-resolved characterization of Ni-MoO2catalysts for the water-gas shift reaction[J].Journal of Physical Chemistry.C,2008,112(6):2121-2128.
[12]KIM Sungho,NAM Sukwoo,LIM Taehoon,et al.Effect of pretreatment on the activity of Ni catalyst for CO removal reaction by water-gas shift and methanation[J].Applied Catalysis B:Environmental,2008,81(1/2):97-104.
[13]CAGLAYAN B S,AKSOYLU A E.Water-gas shift reaction over bimetallic Pt-Ni/Al2O3catalysts[J].Journal of Physical Chemistry,2009,33(10):249-256.
[14]JIANG Lilong,YE Binghuo,WEI Kemei.Effects of CeO2on structure and properties of Ni-Mn-K/bauxite catalysts for water-gas shift reaction[J].Journal of Rare Earths,2008,26(3):352-356.
[15]徐俊,张军营,潘霞,等.CO2矿物碳酸化隔离实验初探[J].化工学报,2006,57(10):2455-2458.XU Jun,ZHANG Junying,PAN Xia,et al.Carbon dioxide sequestration as mineral carbonates[J].Journal of Chemical Industry and Engineering,2006,57(10):2455-2458.
[16]潘霞,张军营,徐俊,等.二氧化碳矿物化隔离研究进展[J].煤炭转化,2006,29(4):78-83.PAN Xia,ZHANG Junying,XU Jun,et al.Research status of carbon dioxide sequestration as mineral carbonation[J].Coal Conversion,2006,29(4):78-83.
猜你喜欢
舰船科学技术(2022年10期)2022-06-17
中国金属通报(2021年2期)2021-12-01
化工时刊(2021年4期)2021-11-02
中国特种设备安全(2020年11期)2020-06-09
上海建材(2020年12期)2020-04-13
电子制作(2019年20期)2019-12-04
当代化工研究(2016年5期)2016-03-20
广州化工(2016年18期)2016-03-12
电源技术(2015年11期)2015-08-22
电力建设(2015年2期)2015-07-12
