
抗肿瘤药物PARP-1抑制剂及其放射性核素标记的研究进展
2011-07-18 01:25:36赵凌舟张华北
同位素 2011年1期
赵凌舟,张华北
(放射性药物教育部重点实验室,北京师范大学 化学学院,北京 100875)
PARP是一种存在于多数真核细胞中的DNA修复酶,最早于1963年由法国斯特拉斯堡大学的Chambon及其研究小组在肝细胞核中发现[1]。这种多功能核酸蛋白通过检测因氧化应激、电离辐射和细胞毒素剂引起的DNA结构损伤片段而被激活[2,3],具有 DNA 损伤修复和维持基因组的稳定性等重要作用,被认为是DNA损伤的感受器。同时,PARP还参与多种蛋白表达的调节、细胞凋亡和坏死、细胞复制和变异的控制,与核糖体蛋白酶活性和蛋白质泛素化作用等有着密 切关系[4-10]。其中,PARP-1与多种恶性肿瘤发生发展的密切关系及其作为肿瘤治疗的新靶点成为近年来研究的热点。
1 PARP-1结构与生理功能
PARP是结构和功能高度保守的蛋白,具有1 014个氨基酸残基,相对分子质量为116 000,由N-末端区、中心自动修复区和C-末端催化区3个功能区组成(PARP-1结构和功能组织示于图1)[11]。N-末端区包括双“锌指”DNA 结合区和核定位信号区,其中双“锌指”DNA结合区能识别DNA缺口并与有缺口的DNA结合。PARP的这种功能是由它的两个特异的“锌指”结构(Zn2+fingers)和核定位信号区 (Nuclear Localization Signal,NLS)决定的。这个区域通过第一和第二“锌指”结构的非序列依赖方式识别双链和单链DNA缺口。C-末端催化区由两部分组成:NAD+结合位点和合成聚腺苷二磷酸核糖的催化位点。中心自动修复区集中了大部分聚ADP核糖化催化位点,被认为是自身聚腺苷二磷酸核糖化的靶位[6,11,12]。
目前PARP家族有18个亚型,其中PARP-1在家族中比例最大,发挥着90%以上的功能,包括介导DNA修复、消耗细胞能量池导致细胞机能障 碍 和 坏 死、促 进 炎 症 基 因 转 录 等[6,7,13]。当DNA损伤出现时,PARP-1迅速被活化,烟酰胺腺嘌呤二核苷酸(NAD+)在PARP-1的催化下分解为烟酰胺和 ADP-核糖(PARP催化NAD+分解的过程示于图2),而ADP-核糖可以聚合在核受体蛋白(主要为PARP-1本身)上,形成线状或直链的PARP-1-ADP核糖多聚物,导致PARP-1的自身修饰,使PARP-1被抑制。这种位阻大、电荷多的多聚物,可以防止周围DNA分子与损伤的DNA进行重组并降低PARP-1与DNA的亲和性,使PAR-1从DNA断裂处解离,然后引导DNA修复酶与DNA缺口结合,对损伤部位进行修复。PARP-1-ADP核糖多聚物从DNA上解离后,经过聚ADP-核糖水解酶[Poly(ADP-ribose)Glycohydrolase,PARG]裂解为ADP-核糖和PARP-1。裂解后的 ADP-核糖可重新与烟酰胺形成NAD+。同时裂解后PRAP-1又重新被激活与DNA结合,如此反复循环进行DNA损伤的修复,最终使细胞存活,PARP的生理功能示于图3。当DNA严重损伤时,超表达的PARP大量消耗 NAD+,引发ATP的耗尽,使细胞处于能量缺乏状态,导致细胞功能紊乱并死亡[14,15]。
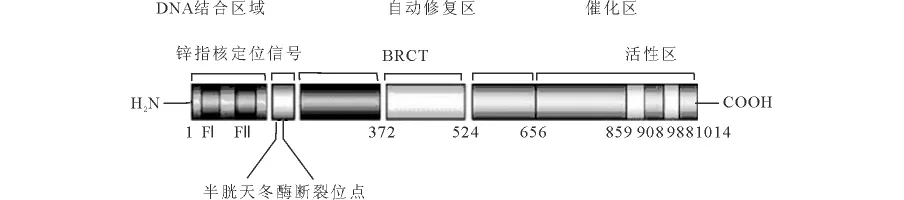
图1 PARP-1结构和功能组织图
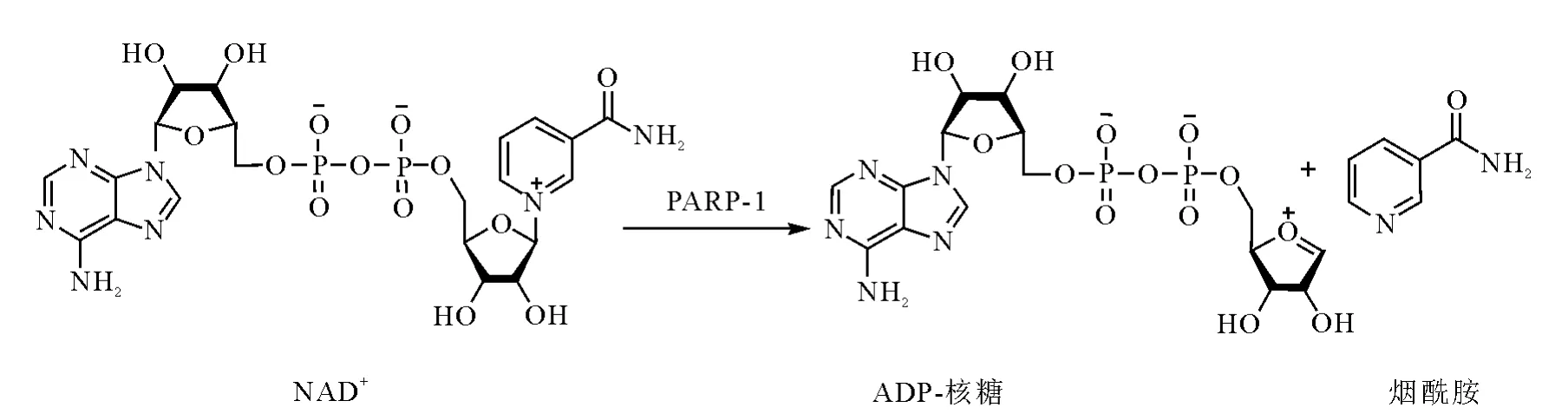
图2 PARP催化NAD+分解的过程
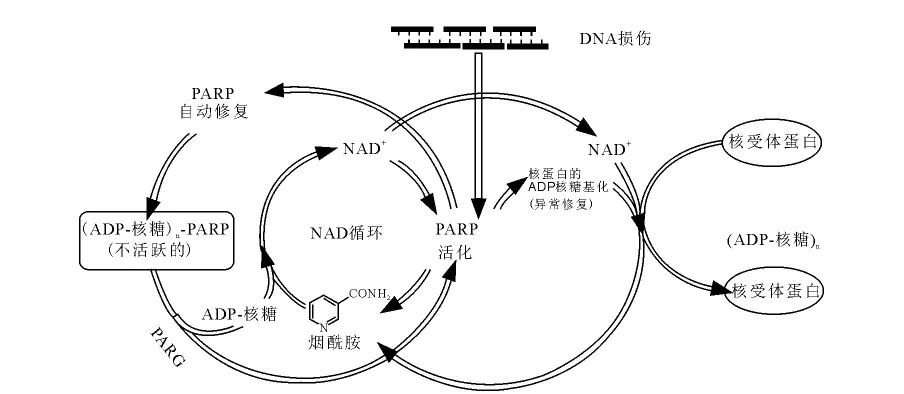
图3 PARP的生理功能
2 PAPR与肿瘤的发生
在维持基因组的稳定性方面,PARP-1的作用至关重要,但抑制PARP-1可能增加机体细胞对烷化剂、电离辐射等DNA损伤因子易感性,导致更容易发生肿瘤。许多研究发现,DNA损伤因子处理敲除PARP-1基因的小鼠可导致肿瘤发生,而单纯PARP-1缺失的小鼠观察至9月龄却未出现任何器官的肿瘤[16]。例如,Nozaki等[17]通过破坏PARP外显子1产生PARP-1缺失(PARP-1-/-)小鼠,再用 AOM(氧化偶氮甲烷)处理PARP-1-/-小鼠后,发现小鼠结肠腺瘤和腺癌、肝脏结节发生率均明显高于PARP-1+/+组,而从出生至9月龄的 PARP-1-/- 小鼠各器官并未发生自发的肿瘤;Tsutsumi等[18]通过破坏 PARP-1外显子1产生 PARP-1-/- 小鼠,然后用BHP(N-二亚硝基-2-羟丙胺)处理小鼠,发现其肝血管瘤、血管肉瘤和肺腺瘤发生率明显高于 PARP-1+/+组;而 Conde等[19]发现,破坏PARP-1外显子4所产生的PARP-1和p53共缺失(PARP-1-/-p53-/-)的小鼠和单纯p53完全缺失(PARP-1+/+p53-/-)组小鼠几乎都死于肿瘤(主要是淋巴瘤、肉瘤、表皮肿瘤和脑肿瘤),然而前组无瘤生存率却明显高于后组。这些似乎表明PARP-1相关的肿瘤的发生存在一定联系。
3 PARP-1抑制剂与肿瘤治疗
由于PARP-1在DNA修复、细胞死亡、增殖分化等方面发挥着重要作用,通过抑制PARP-1活性可抑制PARP-1对DNA修复的机制,从而提高放疗和化疗对肿瘤细胞DNA的损伤,因而PARP-1对肿瘤具有潜在治疗价值。目前,化疗是治疗癌症的主要手段之一。在诸多的化疗药物中,大多是对癌细胞DNA进行破坏来发挥药效,例如替莫唑胺(Temozolomide)使DNA甲基化,喜树碱(Camptothecin)类化合物抑制DNA拓扑异构酶Ⅰ等。但是肿瘤细胞对这类药物经常产生抗药性,其部分原因是由于药物造成的肿瘤细胞DNA损伤的同时,损伤的DNA使核酶PARP大量激活,促进了DNA的修复,因而削弱了药物对癌细胞的杀伤作用。因此,人们开始尝试PARP-1抑制剂与DNA损伤药物(如DNA烷化剂、拓扑异构酶I抑制剂)或放疗联用,以抑制由PARP介导的DNA修复,从而增强治疗效果,并通过减少用药(放射)剂量降低毒副作用。据报道,PARP-1抑制剂能有效增加肿瘤细胞对放疗及烷基化试剂的敏感性。这些结果均提示,PARP-1抑制剂有可能作为肿瘤化疗和放疗的增敏剂,例如,在非毒性剂量下,PARP-1抑制剂可显著提高Topotecan抑制癌细胞生长的活性和Temozolomide的体内抗癌活性[20]。此外,PARP抑制剂能增强肿瘤细胞对N-甲基-N-亚硝脲、博莱霉素、喜树碱、电离放射诱导细 胞 毒 性 的 敏 感 性[21-24]。 在 PARP-1 抑制剂单独使用作为抗肿瘤药物方面,Farmer[25]和Bryant[26]分别报道了PARP-1抑制剂对实验动物的乳腺肿瘤有显著抑制。但是对于PARP-1抑制剂的研究还是重点集中在与DNA损伤药物的联合使用方面。
4 PARP-1抑制剂的研究进展
大部分PARP-1抑制剂为竞争性抑制剂,阻止NAD+与催化区域的结合。已有的X射线衍射结果显示,PARP-1抑制剂主要有以下几个特征:结构中含内酰胺的双环或类似双环的结构(如图4中的A环和B环);含有位于A环上酰胺键另一侧的氢键受体或给体;含有位于A环上酰胺键旁边的疏水性小取代基;含有双环中的疏水性大基团[27]。目前,以PARP-1为靶点的药物研究主要针对肿瘤、神经系统保护和炎症等疾病,尤其是在抗肿瘤方面,PARP-1抑制剂的研究最为广泛。
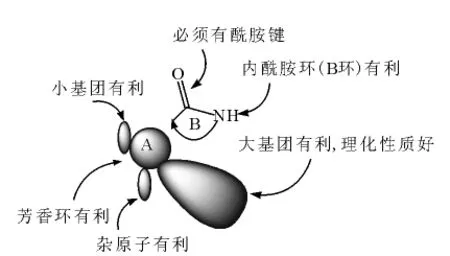
图4 PARP-1抑制剂的结构特征
4.1 传统型PARP-1抑制剂
早期的PARP-1抑制剂是对烟酰胺进行化学修饰而得到。此类抑制剂由于有限的细胞吸收和细胞的滞留时间,其选择性和活性都较差,大部分化合物的水溶性很差[28-29]。烟碱和5-甲基烟碱衍生物是首次见于报道[30]的PARP-1抑制剂。此后,许多结构类似的化合物,例如苯甲酰胺,吡嗪酰胺和苯甲酰胺的取代物,特别是3-氨基苯甲酰胺和3-甲氧基苯甲酰胺(其化学结构示于图5),陆续显示出了对PARP-1存在抑制作用。此后的研究发现,3-氨基苯甲酰胺(3-AB)虽然不是选择性PARP-1抑制剂,但在体外可以阻断各种信号转导途径和介质促炎性上调[31-32],而且体内研究表明,3-AB对各种缺血再灌注损伤器官,例如脑、心、肠、肾和骨骼肌,都有保护作 用[33-38]。 据 报 道[39],苯 甲 酰 胺 衍 生 物 可以抑制葡萄糖的代谢以及DNA和RNA的合成,但对cAMP、磷酸二酯酶、羧肽酶A和胰凝乳蛋白酶的抑制作用弱,能有效地增加肿瘤细胞对放疗及烷基化试剂的敏感性,有可能作为增敏剂联合用于化疗和放疗。
针对3位取代苯甲酰胺类抑制剂的构效关系的研究表明,当3位是供电子基团取代时,活性增强,3位取代苯甲酰胺的活性优于其他位置取代苯甲酰胺的活性[40]。X射线衍射研究表明,PARP-1抑制剂与酶活性部位结合能力的强弱由酰胺键中羰基的构象决定[41]。由于绝大多数PARP-1抑制剂是NAD+的类似物,因此抑制剂和PARP-1的相互作用方式与NAD+类似,即NAD+酰胺上的羰基和C2-C3键之间是反式构象与酶相互作用。PARP-1抑制剂上羰基与PARP-1上的Ser904和QHJ863氨基酸残基也应该形成3个氢键。通过这些研究,设计合成了一系列具有优势构象的双环类PARP-1抑制剂,其化学结构示图6,例如取代的二氢异喹啉-1-酮(2)、酞嗪(2H)-1-酮(3)、喹唑啉-2,4-二酮(4)、取代的喹唑啉-4-酮(5)和二氢苯噁并嗪-4-酮衍生物(6)等,与苯甲酰胺类PARP-1抑制剂相比,这类双环类PARP-1抑制剂的活性更好[42]。
4.2 新型PARP-1抑制剂
新型PARP-1抑制剂大部分是甲酰胺连接芳香环或者氨基甲酰基连接在含有杂原子的多环芳烃,形成芳香类的内酰胺或者酰亚胺,这类化合物与传统的PARP-1抑制剂相比具有更好的活性和选择性。新型PARP-1抑制剂发展于20世纪90年代。Suto[43]和 Banasilk[44]报道了170多个良好的特异性PARP-1抑制剂。并且二氢异喹啉酮类和异喹啉酮类5位置被羟基取代,可以提高活性[45]。后续报道[46-48]显示,5-羟基异喹啉酮(5-AIQ)具有心脏保护功能。Shinkwin等[49]将苯甲酰胺中的苯环用噻吩环取代,设计了一系列新型PARP-1抑制剂,其化学结构示于图7,它们分别是噻吩酰胺衍生物(7)(IC50=8.9μmol/L)、噻吩并嘧啶酮衍生物(8)(IC50=9.7μmol/L)、噻吩并[1,3]恶唑酮(9)及噻吩并吡啶酮衍生物(10)等。初步的活性研究[54]表明,噻吩环取代原来的苯环没有明显降低PARP-1的抑制活性。
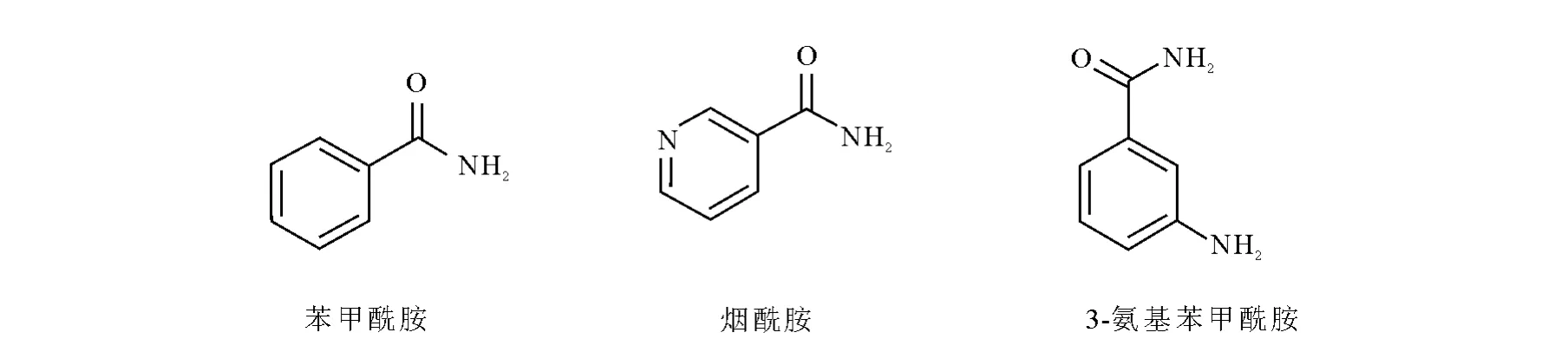
图5 苯甲酰胺、烟碱和3-AB结构式
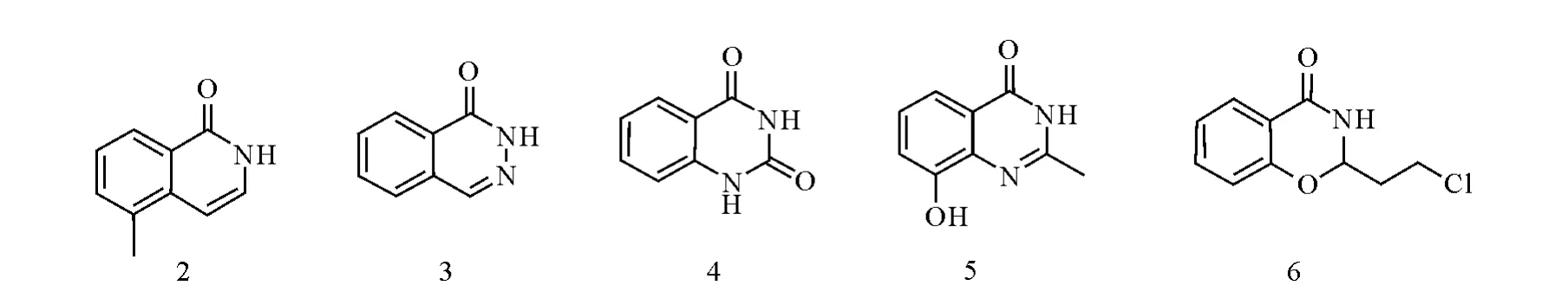
图6 双环类PARP-1抑制剂
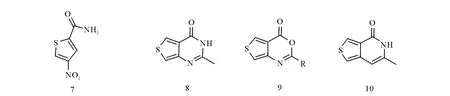
图7 将苯甲酰胺中的苯环用噻吩环取代后形成的一系列新型PARP-1抑制剂
4.3 第三代PARP-1抑制剂
第三代PARP-1抑制剂属于苯并咪唑类、吲哚类和包括一些具有环结构的相关化合物,例如正在用于临床试验的 AG014699[50,51]。这些化合物不仅活性很好、选择性高、溶解性和药物动力学特征有所不同,而且阐述了PARP-1抑制的构效关系[52,53]。PARP-1抑制剂分子需要有空间位置和合适的氢键受体和供体(例如酰胺键中的羰基和N端的活泼H)与PARP-1催化活性位点的Ser-904,Gly-863氨基酸残基结合。N端的活泼H与抑制活性密切相关,倘若N端被甲基化,抑制活性基本消失。此外富电子的芳香环或者杂环平面,与Tyr-907氨基酸残基的π-π相互作用对活性也很关键(PARP-1与其抑制剂的复合物晶体结构结合示意图示于图8)。其中很多化合物是由3-氨基苯甲酰胺的结构演化而来,并具有很好的可溶性和药代动力学性质,例如 正 在 进 行 临 床 试 验 的 ABT-888、BSI-201、AG014699、MK-4827、AZD2281 和 CEP-9722,其相关情况列于表1,其结构式示于图9。虽然这些化合物化学结构和生物利用度各不相同,但都具有较短的半衰期,因此需要频繁给药[54]。其中,BSI-201已于2010年3月通过静脉注射给药与吉西他滨/卡铂联用治疗三阴性乳腺癌的三期临床试验[55],其治疗腹膜癌、子宫癌和卵巢癌的研究也处于Ⅱ期临床阶段,BSI-201很有可能成为第一种通过FDA审批的PARP-1抑制剂[27,56]。
Abbott开发的强效PARP-1抑制剂ABT-888,可以与替莫唑胺、顺铂、卡铂和环磷酰协同作用,明显提升对肿瘤的杀伤作用,且耐受性较好。口服给药后,ABT-888在小鼠、猫、犬体内的生物利用度分别达92%、61% 和72%[57]。ABT-888具有良好的血脑屏障通过性,能够与替莫唑胺联用治疗脑部多形性胶质母细胞瘤,有明显的抑制效果,肿瘤收缩率达67%[58]。目前ABT-888与全脑放疗联合应用治疗转移性脑瘤已进入Ⅰ期临床,而用于转移性乳腺癌、结肠癌和转移性黑色素瘤的治疗也进入Ⅱ期临床阶段。
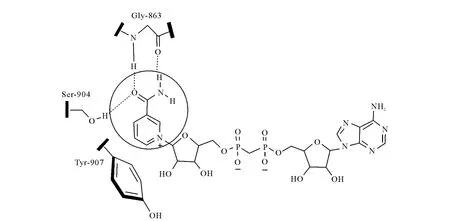
图8 PARP-1与其抑制剂的复合物晶体结构结合示意图

表1 处于临床试验阶段的PARP-1抑制剂
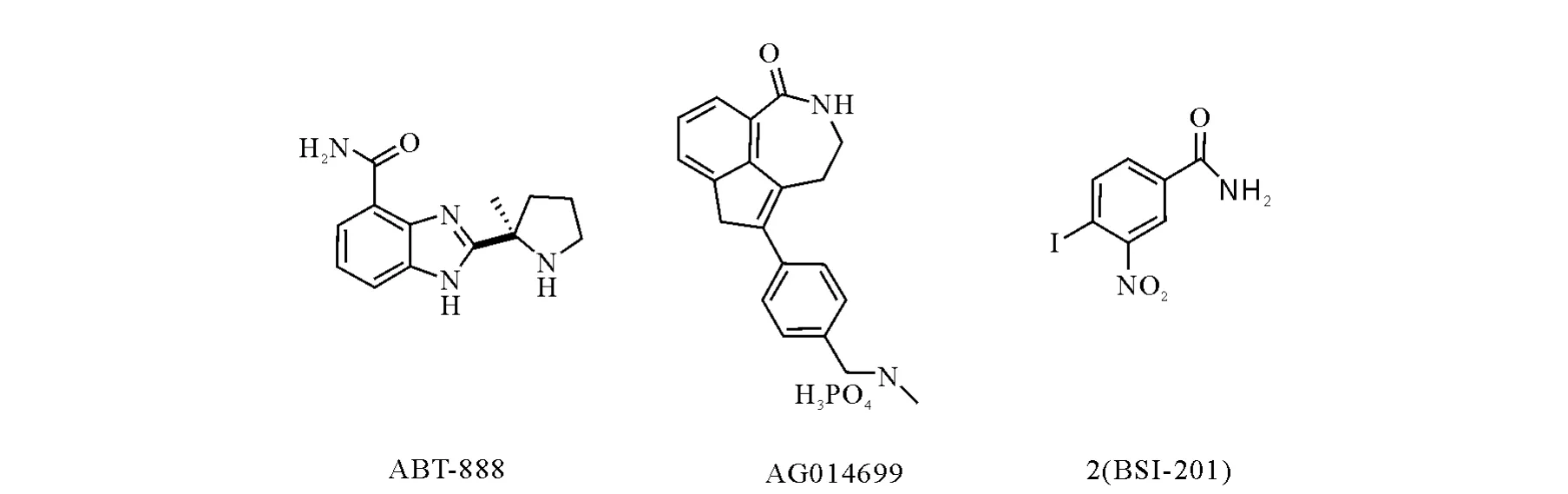
图9 AG014699、ABT-888和2(BSI-201)结构式
2-(4-羟苯基)苯并咪唑-4-酰胺的衍生物,如NU1064(11)、NU1085(12)和 NU1025(13)也是很好的PARP抑制剂,也能加强抗癌药物的细胞毒性作用[31,59,60]。由于 NU1025的溶解度较差,未能进入临床试验,但其对PARP-1的抑制活性是3-AB的50倍。200μmol·L-1浓度的NU1025可使拓扑异构酶Ⅰ抑制剂拓扑替康的抗肿瘤活性增强5倍,使DNA烷化剂替莫唑胺的活性增强6倍,但若单独使用则抗肿瘤活性很弱[61]。通过分子内氢键限制酰胺键构型的化合物NU1085,在浓度只有10μmol·L-1时即可与替莫唑胺发生协同作用,即对肿瘤细胞产生很强的化学致敏效果[62],而且鉴于NU1085合成简单,活性强,且水溶性好,常作为PARP抑制剂的一个对照化合物。NU1085也可以提高拓扑替康的疗效,有可能在癌症治疗中通过联合用药降低肿瘤抗药性和用药剂量。Stacie等基于构效关系和蛋白-配体结构信息设计并合成了一系列吲哚酮类抑制剂(14)[39]。同时,Agouron制药公司的研究组在苯并咪唑甲酰胺类化合物的基础上,以化合物AG014344(15)为基础进一步优化,得到化合物AG014699。该药通过静脉注射给药,作为化疗增强剂与TMZ联用,有很好的抗癌活性,并且在单用或与TMZ联用的情况下都不表现毒性。2003年进入Ⅰ期临床,与替莫唑胺联用用于晚期实体瘤,是第一个进入临床的PARP-1抑制剂。这些化合物的设计符合PARP-1和NAD+结合区域的空间填充和原子间相互作用,其化学结构示于图10。
AZD2281为口服的小分子PARP-1抑制剂,主要用于卵巢癌、乳腺癌和实体瘤的治疗。2009年4月,AstraZeneca报道了AZD2281首次作为单药在Ⅰ期临床中治疗胃癌,其表现出明显抑制效果,耐受性良好,药物毒性95%以上为1~2级。目前该药物与顺铂、卡铂、紫杉醇等药物联用治疗卵巢癌、乳腺癌,实体瘤的研究正处于Ⅱ期临床阶段,预计对于乳腺癌的治疗将于2011年进入Ⅲ期临床。
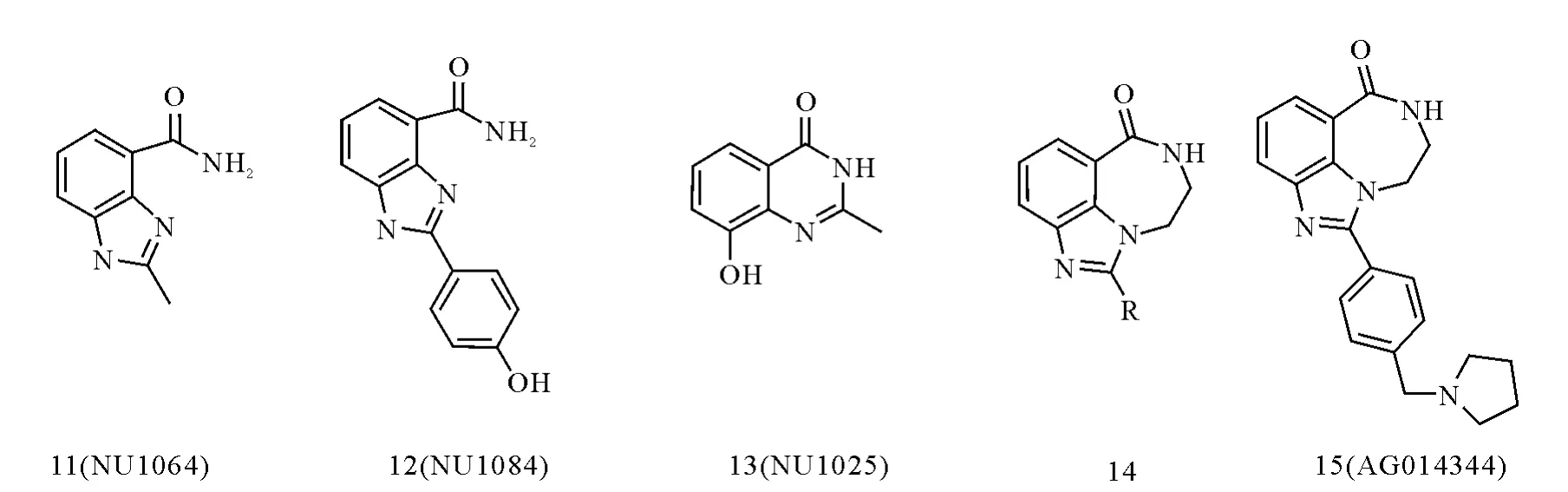
图10 苯并咪唑类和吲哚酮类PARP-1抑制剂

图11 AZD2281、INO1001、INO1188和 Merck4827分子结构式
二氢异喹啉-1-酮和喹唑啉酮类化合物也可以提高抗肿瘤药物对细胞生长的抑制作用。Banasik等[63]报道了一系列5[H]phenanthridin-6-ones取代物的活性比未取代的Phenanthridinone母体活性强100倍。Inotek开发的口服PAPR-1抑制剂药物INO1188(PJ34)抗肿瘤活性良好,可用于治疗脑卒中、类风湿关节炎和再灌注损伤等疾病,但是目前对该药物的研究仍处于临床前研究阶段。该公司开发的另一个静脉注射PARP抑制剂INO1001用于治疗再灌注损伤,正处于Ⅱ期临床阶段,与替莫唑胺联合治疗黑色素瘤也正处于Ⅰ期临床研究阶段[55]。很多PJ43类似化合物的EC50值接近40nmol/L,能够修复各种鼠科动物体外的胸腺细胞[64]。体内模型研究[65]发现,这类化合物可以减少细胞坏死,并改善中风、心肌梗死、糖尿病和关节炎等疾病的症状。
Merck开发的MK4827在人类乳腺癌细胞系中的IC50为3.8nmol·L-1,口服给药后,在猫、犬、猴体内的生物利用度分别达65%、80%和70%。MK4827与卡铂联用治疗晚期实体瘤和卵巢癌,目前正处于Ⅰ期临床研究阶段[66]。
美国Cepkalon公司采用高通量筛选的方法,发现了一种全新的PARP-1抑制剂,其母核为吡咯并咔唑内酰胺。化合物16的活性好,其余的衍生物的活性都较低。后续报道的化合物CEP-6800(17),不仅水溶性较好,而且显著增加了对PC12细胞的毒性,并在体外对多种肿瘤细胞系显示了增强TMZ、依立替康和顺铂等药物的抗癌活性的作用[67]。但由于CEP-6800存在骨髓毒性,未能进入临床试验阶段。经过对CER-6800进一步修饰,得到化合物 CEP-8983(18),虽然解决了骨髓毒性的问题,但水溶性较CER6800差。为增加CEP-8983的水溶性,在其酰胺的氮原子上引入二烷基氨甲基得到化合物18的前药CEP-9722(19)。2009年5月开始进入三期临床试验阶段,主要针对实体瘤的治疗。吡咯并咔唑内酰胺类PARP-1抑制剂化学结构示于图12。
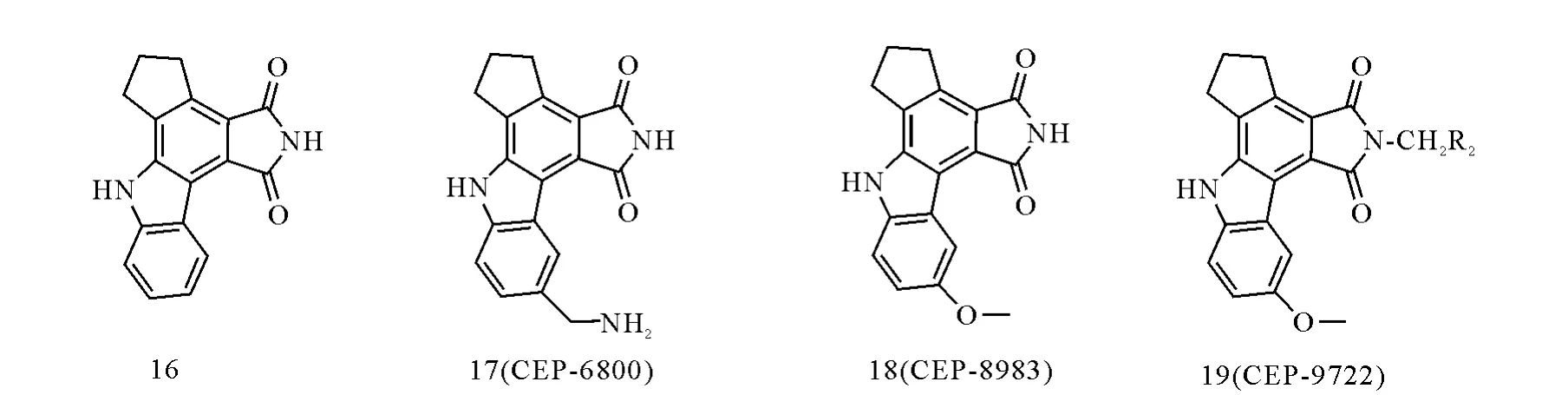
图12 吡咯并咔唑内酰胺类PARP-1抑制剂
5 放射性核素标记PRAP-1抑制剂的研究进展
PET是将发射正电子的核素标记物引入体内,采集数据通过计算机重建图形得到器官断层影像的一种新技术,分辨能力较单光子发射计算机断层(SPECT)明显提高;且由于其所用正电子核素大多是构成人体的基本元素或其类似物,如11C、13N、15O、18F等,标记物多是人体生理物质,如葡萄糖、水、氨基酸和神经介质等,故PET图像实质上是反映某种生理物质在人体内的动态变化和代谢过程,是在分子水平上反映人体的生理或病理变化。PET是人类第一次在活体分子水平上完成生物学显示的显像技术,可以在器官尚未发生结构变化的阶段对疾病进行早期诊断。
关于用放射性核素标记PARP-1抑制剂的研究,最早是在1994年,Andersson等[68]报道了11C标记的苯甲酰胺类化合物,作为聚(ADP-核糖)合成酶的示踪剂。11C均标记在3-氨基苯甲酰胺、3-甲氧基苯甲酰胺和烟酰胺的酰胺基上,其合成路线示于图13。其反应时间约为5 min,标记的产率为45%~70%,放化纯度均大于99%,放射性比活度为2~3μCi·mol-1。在猴子体内的分布和药代动力学表明,11C标记的化合物在肝、肾和淋巴组织中摄取高,而且存在滞留情况,其中11C烟碱虽然血清除快,但相对于11C标记的3-氨基苯甲酰胺和3-甲氧基苯甲酰胺慢。除了11C标记的3-甲氧基苯甲酰胺起始脑摄取较高外,11C标记的烟碱和3-氨基苯甲酰胺脑摄取低,而且清除均太快。给药3min后的显像表明,虽然有部分进脑,但肝肾摄取过高,显像效果并不理想,而且也并没有证明标记的化合物与PARP存在很好的结合。
2000年,Yoshinori Miyake等[69]报道了11C标记 的 异 喹 啉 类 化 合 物 (11C-MIQO,3,4-dihydro-5-[11C]methoxy-1(2H)-isoquinolinone),作为聚(ADP-核糖)合成酶的示踪剂,检测PARP-1的过度表达。图14为11C-MIQO 的合成过程。其反应时间较长,约为35min,标记率较低,小于40%,放化纯度大于99%,比活度76GB·qμmol-1。11C-MIQO 在老鼠和猴子脑中药代动力学研究表明,尽管正常小鼠的11CMIQO脑摄取低,但是11C-MIQO进脑速度快,脑清除也快,且其分布结果不同于[15O]H2OPET得到的脑血流分布结果,这主要与11CMIQO在脑中的快速清除有关。虽然在猴脑周围的肌肉组织中发现了少量的11C-MIQO滞留,但不影响利用PET检测猴脑中缺血性损伤区域对11C-MIQO的摄取,这主要是因为缺血性损伤增加了PARP抑制剂能够结合的活化的NAD结合位点。聚(ADP-核糖)的浓度可以反映PARP酶活性,当完整细胞用氧化剂处理后,聚(ADP-核糖)的浓度迅速增加,然后用0.01 mmol/L 1,5-dihydroisoquinolinone(MIQO 类似物)处理细胞,发现95%的由氧化剂诱发的聚(ADP-核糖)减少了。另外,小鼠的局灶性脑缺血模型体内实验表明,3,4-dihydro 5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone能 够 大幅度减少梗塞面积。这些结果意味着,PARP抑制剂仅仅在有DNA损伤的细胞处积累,并且与DNA的损伤程度相关。这些结果主要与在正常组织中11C-MIQO的快速进入和快速清除性质有关。11C-MIQO有可能成为一种示踪剂,利用PET检测缺血性损伤区域的PARP过度表达,但是还需要进一步的实验,以证实11C-MIQO在缺血性损伤的组织中具有特异性积累。
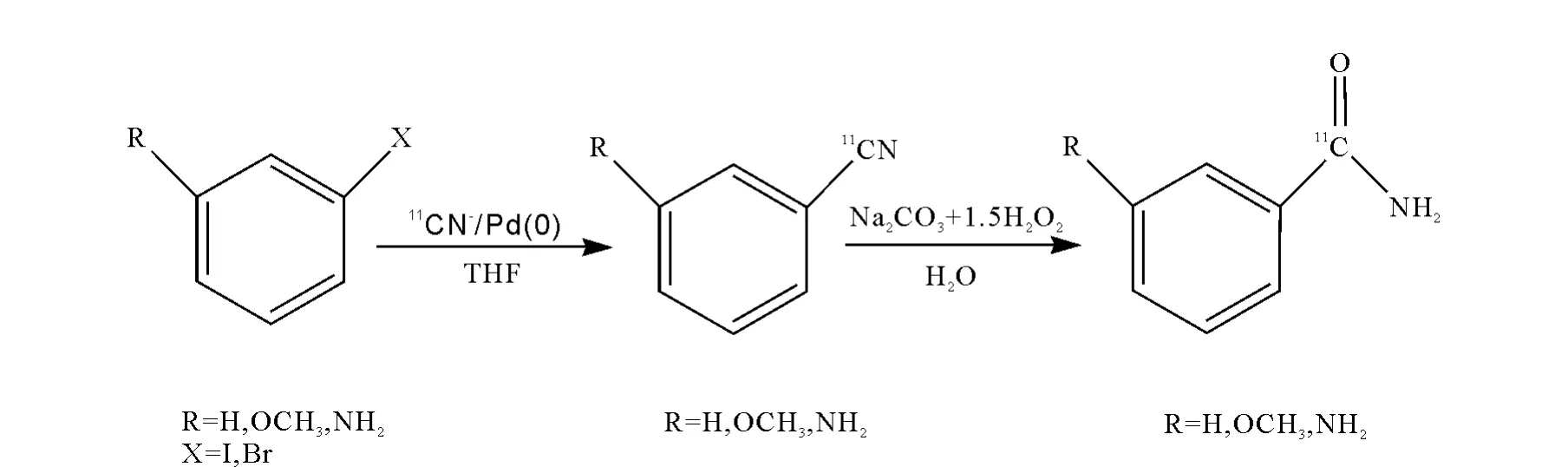
图13 11C标记的苯甲酰胺类化合物合成路线
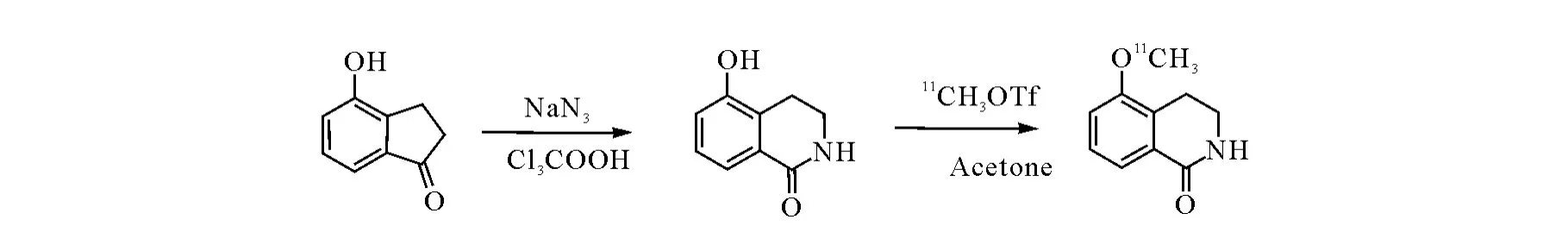
图14 11C-MIQO的合成步骤
2005 年,Zhude Tu 等[70]报 道 了 Phenanthridinone类衍生物,2-(dimethylamino)-N-(5,6-dihydro-6-oxophenanthridin-2-yl)acetamide,11C-PJ34作为细胞凋亡PARP-1的显像剂,与PARP-1具有很好的结合关系,IC50值达到了nm级,11C-PJ34的合成和标记过程示于图15。其反应时间比较长,在50min以上,标记率约为60%,放化纯度大于99%,比活度接近740Bq/mol。静脉注射高剂量的含有葡萄糖的亚硝基脲(STZ),进入到细胞的STZ干扰主要在β细胞表达的转运葡萄糖的GLUT-2,破坏胰腺的β细胞导致Ⅰ型糖尿病产生。由于肝细胞也表达GLUT-2,所以肝细胞中的DNA也会被STZ损伤。其动物分布实验表明:与对照组相比,接收STZ剂量的动物组中,胰腺和肝对11C-PJ34有更高的摄取。而且,缺少GLUT-2的组织对11CPJ34的摄取也没有区别。免疫组学研究也表明,与对照组相比,在用STZ处理的动物的胰腺β细胞中存在大量的利用免疫组学染色的PAR。这些结果进一步证实了用STZ处理动物后,其胰腺和肝对11C-PJ34有更高的摄取,也表明了11C-PJ34能够反映在早期的细胞坏死阶段、PARP-1活性的过度表达。
2009年Riss等[71]报道了18F标记的哌仑西平和其代谢物 N5-18F氟代乙酯-LS 75(化合物4,18F-FE-LS-75)及类似物化合物3(图16)的研究。其反应时间在2h左右,标记率也较低,只有约30%,放化纯度大于98%,比活度67~89 GBq·μmol-1。其中化合物3的进脑虽然较化合物4好,但是活性低(大于3μmol/L),代谢慢,不能成为PARP-1的示踪剂。而化合物4体外实验稳定性好,对PARP-1的抑制活性中等(0.2μmol/L),进脑量较好,有潜力成为一种PARP-1的PET显像剂。
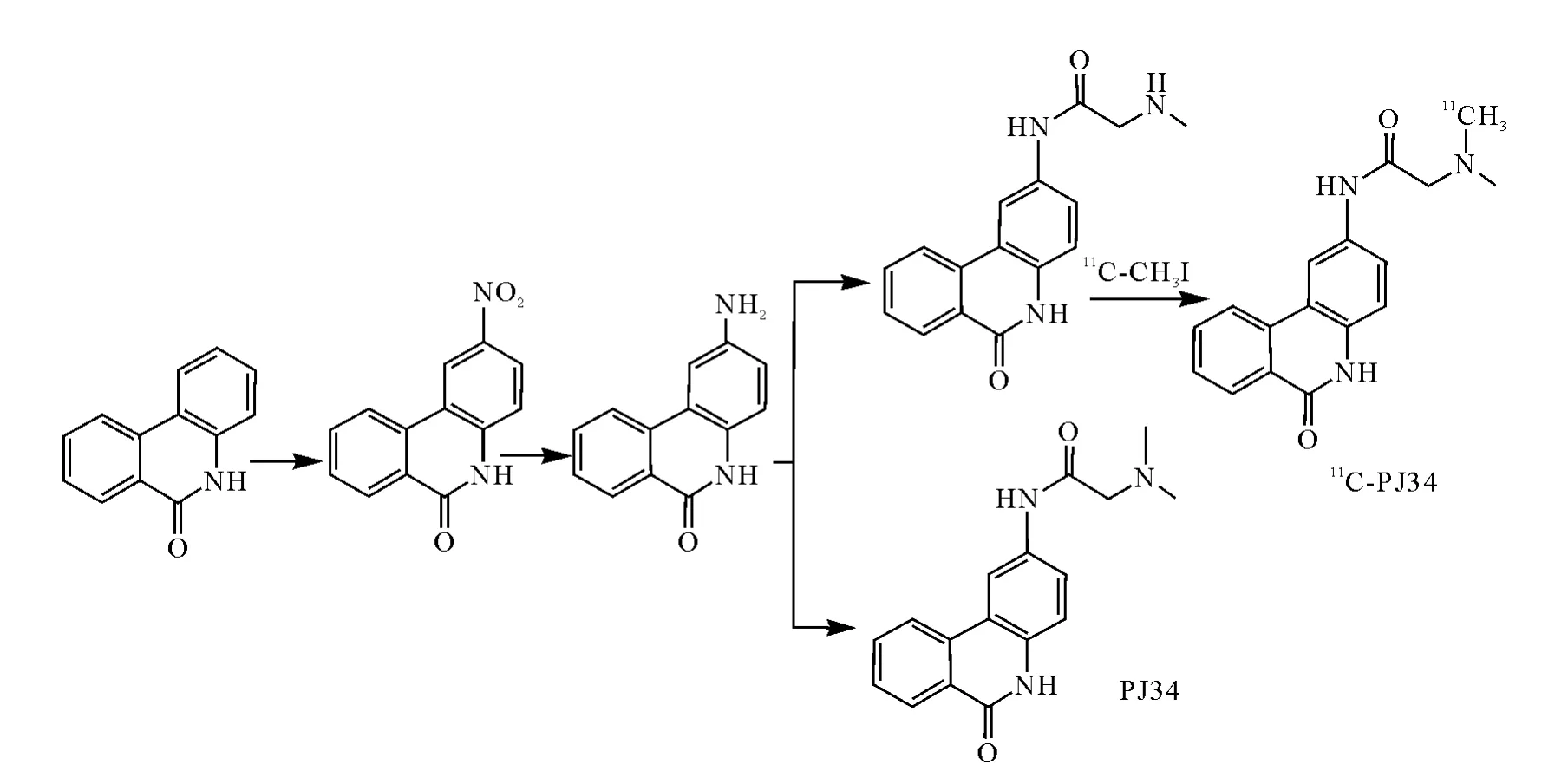
图15 11C-PJ34的合成和标记路线
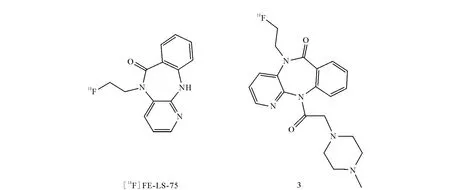
图16 18F-FE-LS-75和化合物3的结构式
5 小结与展望
在过去的几十年中,研究人员已经对PARP-1的结构和PARP-1抑制剂在治疗一些重大疾病中的作用进行了广泛而深入的研究,迄今为止,尽管还没有PARP-1抑制剂作为药物上市,但越来越多的PARP-1抑制剂分子受到关注,尤其是在三阴性乳腺癌和晚期卵巢癌等难治癌症治疗上的成果引人注目。随着人们对PARP家族中其他成员的深入研究,凭借在PARP-1抑制剂研究已经取得的成果,其他成员也会成为新的潜在药物作用靶点,进而开辟新的抗肿瘤药物研究领域。在放射性核素标记PARP-1抑制剂的研究中,主要标记核素是11C和18F,通过PET检测PARP-1的过度活化,早期诊断肿瘤的发生和发展。目前虽然报道的较少,但是随着PARP-1抑制剂的不断深入,新的PARP-1抑制剂的不断出现,放射性核素标记PARP-1抑制剂的研究也会越来越丰富,在肿瘤早期诊断和治疗领域也会占有一席之地。
[1] Chambon P,Weill JD,Mandel P.Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme[J].Biochem Biophys Res Commun,1963,11(1):39-43.
[2] Bai P,Bakondi E,Szabo EE,et al.Partial protection by poly(ADP-ribose)polymerase inhibitors from nitroxyl-induced cytotoxity in thymocytes[J].Free Radic Biol Med,2001,31:1 616-1 623.
[3] Burkle A.Physiology and pathophysiology of poly(ADP-ribosyl)ation[J].Bioessays,2001,23:795-806.
[4] Shall S,de Murcia G.Poly(ADP-ribose)polymerase-1,what have we learned from the deficient mouse model[J].Mutat Res,2000,460:1-15.
[5] Kim MY,Mauro S,Gervy N,et al.NAD+dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1[J].Cell,2004,119:803-814.
[6] Kim MY,Zhang T,Kraus WL.Poly(ADP-ribosyl)ation by PARP-1:PAR-laying NAD+into a nuclear signal[J].Genes Dev,2005,19:1 951-1 967.
[7] Schreiber V,Danter F,Ame JC,et al.Poly(ADP-ribose):novel functions for an old molecule[J].Nat Rev Mol Cell Biol,2006,7:517-528.
[8] Saxena A,Saffery R,Wong LH,et al.Centromere proteins Cenpa,Cenpb,and Bub3interact with poly(ADP-ribose)polymerase-1protein and are poly(ADP-ribosyl)ated[J].J Biol Chem,2002,277:26 921-26 926.
[9] Samper E,Goytisolo FA,Menissier de Murcia J,et al.Normal telomere length and chromosomal end capping in poly(ADP-ribose)polymerase-deficient mice and primary cells despite increased chromosomal instability[J].J Cell Biol,2001,154:49-60.
[10]Burkle A.Poly(ADP-ribose):the most elaborate metabolite of NAD+[J].FEBS J,2005,44:4 576-4 589.
[11]Sodhi RK,Nirmal Singh,Jaggi AS.Poly(ADP-ribose)polymerase-1 (PARP-1)and its therapeutic implications[J].Vascular Pharmacology,2010,53:77-87.
[12]Alvarez Gonzalez R,Pacheco Rodriguez G,Mendoza Alvarez H.Eazymology of ADP-ribose polymer synthesis[J].Mol Cell Biochem,1994,138(1-2):33-37.
[13]Ame JC,Spenlehauer C,de Murica G.The PARP superfamily[J].Bioessays,2004,26:882-893.
[14]Berger NA.Poly(ADP-ribose)in the cellular response to DNA damage[J].Radiat Res,1985,101(1):4-15.
[15]Virag L,Szabo C.The therapeutic potential of poly(ADP-ribose)polymerase inhibitors[J].2002,54:375-429.
[16]Nozaki T,Fujihara H,Watanabe M,et al.Parp-1 deficiency implicated in colon and liver tumorigenesis induced by azoxymethane[J].Cancer Sci,2003,94:497-500.
[17]Nozaki T,Fujihara H,Watanabe M,et al.Parp-1 deficiency implicated in colon and liver tumorigenesis induced by azoxymethane[J].Cancer Sci,2003,94:497-500.
[18]Tsutsumi M,Masutani M,Nozaki T,et al.Increased susceptibility of poly(ADPribose)polymerase-1knockout mice to nitrosamine carcinogenicity[J].Carcinogenesis,2001,22:1-3.
[19]Conde C,Mark M,Oliver FJ,et al.Loss of poly(ADP-ribose) polymerase-1causes increased tumour latency in p53-deficient mice[J].EMBO J,2001,20:3 535-3 543.
[20]Calabrese CR,Batey MA,Thomas HD,et al.I-dentification of potent nontoxic poly(ADP-ribose)polymerase-1inhibitors:chemopotentiation and pharmacological studies[J].Clin Cancer Res,2003,9:2 711-2 718.
[21]Miki K,Uehara N,Shikata N,et a1.Poly(ADP-ribose)polymerase inhibitor 3-aminobenzamide rescues N-methyl-N-nitrosourea induced photoreceptor cell apoptosis in sprague-dawley rats through preservation of nuclear factor—kappaB activity[J].Exp Eye Res,2007,84,2:285-292.
[22]Rose JL,Reeves KC,Likhotvorik RI,et al.Base excision repair proteins are required for integrinmediated suppression of bleomycin-induced DNA breakage in murine lung endothelial cells[J].J Pharmacol Exp Ther,2007,321,1:318-326.
[23]Thomas HD,Calabrese CR,Batey MA,et a1.Preclinicel selection of a novel poly(ADP-ribose)polymerase inhibitor for clinical trial[J]. Mol Cancer Ther,2007,6(3):945-956.
[24]Dungey FA,Löser DA,Chalmeers AJ.Replication-dependent radio-sensitization of human glioma cells by inhibition of poly(ADP-ribose)polymerase:mechanisms and therapeutic potential[J].Int J Radiat Oncol Biol Phys,2008,72,4:1 188-1 197.
[25]Farmer H,McCabe N,Lord CJ,et al.Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy[J].Nature,2005,434:917-921.
[26]Bryant HE,Schultz N,Thomas HD,et al.Specific killing of BRCA2deficient tumours with inhibitors of poly(ADP-ribose)polymerase[J].Nature,2005,434:913-917.
[27]张可辉,张亮仁,张礼和.PARP-1抑制剂在抗肿瘤方面的研究进展[J].中国药学杂志,2010,45(22):1 689-1 694.
[28]Gabriele Costantino,Antonio Macchiarulo,Emidio Camaioni,et al.Modeling of poly(ADP-ribose)polymerase(PARP)inhibitors:Docking of ligands and quantitative structure-activity relationship analysis[J].J Med Chem,2001,44:3 786-3 794.
[29]Watson CY,Whishb WJD,Threadgill MD.Synthesis of 3-substituted benzamides and 5-substituted isoquinolin-1(2H)-ones and preliminary evaluation as inhibitors of poly(ADP-ribose)polymerase(PARP) [J].Bioorg Med Chem,1998,6:721-734.
[30]Clark JB,Ferris JM,Pinder S.Control of nucleic acid and nicotinamide nucleotide synthesis in generating rat liver[J].Bichem Biophys Acta,1971,238:82-87.
[31]Cosi C.New inhibitors of poly(ADP-ribose)polymerase and their potential therapeutic targets[J].Expert Opin Ther Patents,2002,12:1 047-1 071.
[32]Watson CY,Whish WJ,Threadgill MD.Synthesis of 3-substituted benzamides and 5-substituted isoquinolin-1(2H)-ones and preliminary evaluation as inhibitors of poly(ADP-ribose)polymerase(PARP)[J].Bio Org Med Chem,1998,6:721-734.
[33]Bowes J,McDonald MC,Piper J,et al.Inhibitors of poly(ADP-ribose)synthetase protect rat cardiomyocytes against oxidant stress[J].Cardiovasc Res,1999,41:126-134.
[34]Liaudet L,Szabo E,Timashpolsky L,et al.Suppression of poly(ADP-ribose)polymerase activation by 3-aminobenzamide in a rat model of myocardial infarction,long-term morphological and functional consequences[J].Br J Pharmacol,2001,133:1 424-1 430.
[35]Liaudet L,Szabo A,Soriano FG,et al.Poly(ADP-ribose)synthetase mediates intestinal mucosal barrier dysfunction after mesenteric ischemia[J].Shock,2000,14:134-141.
[36]Cuzzocrea S,Zingarelli B,Costantino G,et al.Beneficial effects of 3-aminobenzamide,an inhibitor of poly(ADP-ribose)synthetase in a rat model of splanchnic artery occlusion and reperfusion[J].Br J Pharmacol,1997,121:1 065-1 074.
[37]Chatterjee PK,Cuzzocrea S,Thiemermann C.Inhibitors of poly(ADP-ribose)synthetase protect rat proximal tubular cells against oxidant stress[J].Kidney Int,1999,56:973-984.
[38]Thiemermann C,Bowes J,Myint F,et al.Inhibition of the activity of poly(ADP-ribose)synthetase reduces ischemia-reperfusion injury in the heart and skeletal muscle[J].Proc Nat Acad Sci,1997,94:679-683.
[39]Griffin RJ,Curtin NJ,Newell DR,et al.The role of inhibitors of poly(ADP-ribose)polymerase as resistance-modifying agents in cancer therapy[J].Biochimie,1995,77:408-422.
[40]Watson CY,William JDW,Threadgill MD.Threadgill:synthesis of 3-substituted benzamides and 5-substituted isoquinolin-1(2H)-ones and preliminary evaluation as inhibitors of poly(ADP-ribose)polymerase (PARP)[J].Bioorg Med Chem,1998,6:721-734.
[41]Li Hong,Goldstein BM.Carboxamide group conformation in the icotinamide and thiazole-4-carboxamide rings:implications for enzyme binding[J].J Med Chem,1992,35:3 560-3 567.
[42]沈超,吴晓明,孙宏斌.聚腺苷二磷酸核糖聚合酶抑制剂[J].药物进展,2006,30(1):5-11.
[43]Banasik M,Komura H,Shimoyama M,et al.Specific inhibitors of poly(ADP-ribose)synthetase and mono(ADP-ribosyl)transferase[J].J Biol Chem,1992,267:1 569-1 575.
[44]Suto MJ,Turner WR,Werbel LM,et al.Dihyrdroisoquinolinones:the design and synthesis of a new series of potent inhibitors of poly(ADP-ribose)polymerase[J].Anticancer Drug Dev,1991,6:107-117.
[45]Southan GJ,Szabo C.Poly(ADP-ribose)polymerase inhibitors[J].Curr Med Chem,2003,10:321-340.
[46]Docherty JC,Kuzio B,Silvester JA,et al.An inhibitor of poly(ADP-ribose)synthetase activity reduces contractile dysfunction and preserves high energy phosphate levelsduring reperfusion of the ischaemic rat heart[J].Br J Pharmacol,1999,127:1 518-1 524.
[47]McDonald MC,Mota-Filipe H,Wright JA,et al.Effects of 5-aminoisoquinolinone,a water-soluble,potent inhibitor of the activity of poly(ADP-ribose)polymerase on the organ injury and dysfunction caused by haemorrhagic shock[J].Br J Pharmacol,2001,130:843-850.
[48]Wayman N,McDonald MC,Thompson AS,et al.5-Aminoisoquinolinone,apotent inhibitor of poly(adenosine 5’-diphosphateribose)polymerase,reduces myocardial infarct size[J].Eur J Pharmacol,2001,430:93-100.
[49]Shinkwin AE,William JDW,Threadgill MD.Synthesis of thioph-enecarboxamides,thieno[3,4-c]pyridin-4(5H)-ones and thieno[3,4-d]pyrimidin-4(3H)-ones and preliminary evaluation as inhibitors of poly(ADP-ribose)polymerase(PARP)[J].Bioorg Med Chem,1999,7:297-308.
[50]Canan Koch SS,Thoresen LH,Tikhe JG,et al.Novel tricyclic poly(ADP-ribose)polymerase-1inhibitors with potent anticancer chemopotentiating activity:design,synthesis,and X-ray cocrystal structure[J].J Med Chem,2002,45:4 961-4 974.
[51]Plummer R,Jones C,Middleton M,et al.PhaseⅠ study of the poly(ADP-ribose)polymerase inhibitor,AG014699,in combination with temozolomide in patients with advanced solid tumors[J].Clin Cancer Res,2008,14:7 917-7 923.
[52]Penning TD,Zhu GD,Gandhi VB,et al.Discovery and SAR of 2-(1-propylpiperidin-4-yl)-1H-benzimidazole-4-carboxamide:apotent inhibitor of poly(ADP-ribose)polymerase(PARP)for the treatment of cancer[J].Bioorg Med Chem,2008,16,14:6 965-6 975.
[53]Penning TD,Zhu GD,Gandhi VB,et al.Discovery of the poly(ADP-ribose)polymerase(PARP)inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide(ABT-888)for the treat-ment of cancer[J].J Med Chem,2009,2(2):514-523.
[54]Donawho CK,Luo Y,Luo YP,et al.ABT-888,an orally active poly(ADP-ribose)polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models[J].Clin Cancer Res,2007,13:2 728-2 737.
[55]Rodon J,Iniesta MD,Papadopoulos K.Development of PARP inhibitors in oncology[J].Expert Opin Investig Drugs,2009,18(1):31-43.
[56]Ferraris DV.Evolution of poly(ADP-ribose)polymerase-1 (PARP-1)inhibitors from concept to clinic[J].J Med Chem,2010,53:4 561-4 584.
[57]Donawho CK,Luo Y,Penning TD,et al.ABT-888,an orally active poly(ADP-ribose)polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models[J].Clin Cancer Res,2007,13(9):2 728-2 737.
[58]Fauzee NJ,PAN J,Wang YL.PARP and PARG inhibitors:New therapeutic targets in cancer treatment[J].Pathol Oncol Res,16,4:469-478.
[59]Delaney CA,Green MH,Lowe JE,et al.Endogenous nitric oxide induced by interleukin-1beta in rat islets of Langerhans and HIT-T15cells causes significant DNA damage as measured by the“comet”assay[J].FEBS Lett,1993,333:291-295.
[60]White AW,Almassy R,Calvert AH,et al.Resistance-modifying agents:Synthesis and biological properties of benzimidazole inhibitors of the DNA repair enzyme poly(ADP-ribose)polymerase[J].J Med Chem,2000,43:4 084-4 097.
[61]Cepeda V,Fuertes MA,Castilla J,et al.Poly(ADP-ribose)polymerase-1(PARP-1)inhibitors in cancer chemotherapy[J].Recent Pat Anticancer Drug Discov,2006,1(1):39-53.
[62]Curtin NJ.PARP inhibitors for cancer therapy[J].Expert Rev Mol Med,2005,7(4):1-20.
[63]Jagtap P,Szabo C.Poly(ADP-ribose)polymerase and the therapeutic effects of its inhibitiors[J].Nat Rev Drug Discov,2005,4:421-440.
[64]Haddad M,Rhinn H,Bloquel C,et al.Antiinflammatory effects of PJ34,apoly(ADP-ribose)polymerase inhibitor,in transient focal cerebral ischemia in mice[J].Br J Pharmacol,2006,149:23-30.
[65]Hamby AM,Suh SW,Kauppinen TM,et al.Use of a poly(ADP-ribose)polymerase inhibitor to suppress inflammation and neuronal death after ischemia-reperfusion [J]. Stroke, 2007, 38:632-636.
[66]Jones P.Development of MK-4827:A novel oral poly(ADP-ribose)polymerase(PARP)inhibitor efficacious in BRCA-1and-2mutant tumors[C]//238th National Meeting of the American Chemical Society,2009.
[67]Miknyoczki SJ,Jones-Bolin S,Pritchard S,et al.Chemopotentiation of temozolomide,irinotecan,and cisplatin activity by CEP-6800,apoly(ADP-ribose)polymerase inhibitor[J].Mol Cancer T-her,2003,2:371-382.
[68]Andersson Y,Bergstrom M,Langstrom B.Synthesis of11C-labelled benzamide compounds as potential tracers for poly(ADP-ribose)synthetase[J].Appl Radiat Isot,1994,45:707-714.
[69]Miyake Y,Kuge Y,Shimadzu H,et al.Biodistribution of 3,4-dihydro-5-[11C]methoxy-1(2H)-isoquinolinone,apotential PET tracer for poly(ADP-ribose)synthetase[J].Nuclear Medicine &Biology,2000,27:701-705.
[70]Tu Zhude,Chu Wenhua,Zhang Jun,et al.Synthesis and in vivo evaluation of[11C]PJ34,apotential radiotracer for imaging the role of PARP-1 in necrosis[J].Nuclear Medicine & Biology,2005,32:437-443.
[71]Riss PJ,Soskic V,Schrattenholz A,et al.Synthesis and radiosynthesis of N5-[18F]fluoroethylpirenzepine and its metabolite N5-[18F]fluoroethyl-LS 75[J].J Label Compd Radiopharm,2009,52:576-579.
猜你喜欢
心肺血管病杂志(2020年3期)2021-01-14 00:42:34
上海农业学报(2017年3期)2017-04-10 12:39:26
中国生化药物杂志(2016年5期)2016-07-24 17:30:25
现代农业(2016年4期)2016-02-28 18:42:14
中国当代医药(2015年16期)2015-03-01 02:03:13
应用化工(2014年1期)2014-08-16 13:34:08
中国药理学通报(2014年2期)2014-05-09 08:22:39
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:21
化工生产与技术(2014年3期)2014-02-27 13:41:44
金属矿山(2013年11期)2013-03-11 16:55:05
