
膜性肾病合并Alport综合征
2011-06-24 14:42曾彩虹刘志红
肾脏病与透析肾移植杂志 2011年2期
黄 倩 曾彩虹 刘志红
·肾活检·
膜性肾病合并Alport综合征
黄 倩 曾彩虹 刘志红
报道1例中年男性患者临床为肾病综合征伴血压升高,无听力及视力的下降,无肾脏病家族史;肾活检光镜及常规免疫荧光符合肾小球膜性病变,但肾间质见簇状分布的泡沫细胞,进一步行肾组织IV型胶原染色及电镜检查,最终明确合并Alport综合征。
膜性肾病 Alport综合征 肾活检
病例摘要
病史 38岁男性患者,因“水肿、尿检异常5年”于2008-07-28入院。
缘于2003年6月无明显诱因出现双下肢及颜面水肿,检查发现尿蛋白4+,隐血阴性,血白蛋白减低(具体不详),肾功能正常,血压升高(最高达190/100 mmHg),予泼尼松60 mg/d治疗1月,尿检无缓解;遂予甲泼尼龙冲击(共3.5g)及环磷酰胺冲击(0.8g×3次),后续以泼尼松60 mg/d连续服4月后逐渐减量,期间同时服用中药汤剂治疗1年余,尿检持续无缓解。2006年曾在外院行肾活检术,诊断“膜性肾小球肾炎(II~III期)”,再次予甲泼尼龙片40 mg/d,霉酚酸酯1.5 g/d及贝那普利、缬沙坦、美托洛尔降压治疗,血压控制在130/80 mmHg左右,1年停霉酚酸酯,甲泼尼龙片逐渐减量;至2008年4月停服;期间曾加服雷公藤多苷90 mg/d治疗,尿蛋白无缓解,波动于1+~4+。病程中患者精神、食欲正常,体重减轻10 kg。
既往史、家族史:无特殊。
体格检查 体温36.7℃,脉搏100次/min,呼吸20次/min,血压120/80 mmHg,BMI 23.98 kg/m2。发育正常,营养良好,双侧眼睑无水肿,角膜透明,瞳孔对光反射灵敏,听力无下降,心、肺未及异常,腹平软,全腹无压痛及反跳痛,肝、脾肋下未及,肝、肾区无叩痛,腹部移动性浊音(+),肠鸣音不亢,双下肢轻度凹陷性水肿。
实验室检查
尿液 尿蛋白2.47~11.99 g/24 h,尿沉渣红细胞1万/ml,α2-MG>32 mg/L,C3>32 mg/L;视黄醇结合蛋白(RBP)1.0 mg/L,NAG酶84.9 U/g·cr,溶菌酶(Lyso)1.19 mg/L,尿渗量567 mOsm/kg·H2O。
血常规 Hb 131 g/L,WBC 7.8×109/L,PLT 279×109/L。
血生化 Alb 25.1 g/L,Glo 14.1 g/L,BUN 2.06 mmol/L,SCr59.23μmol/L,UA 338μmol/L,胱抑素C 1.07 mg/L,ALT 18 U/L,AST 17 U/L,总胆固醇6.44mmol/L,三酰甘油3.21 mmol/L,K+3.01 mmol/L,Na+143.1 mmol/L,Cl-108.3 mmol/L,TCO227.7 mmol/L,Ca2+1.9 mmol/l,P3+1.24 mmol/L。C反应蛋白3.1 mg/L。空腹血糖4.52 mmol/L,餐后2h血糖6.1 mmol/L。
免疫学 IgG 3.69 g/L,IgA 1.41 g/L,IgM 0.445 g/L,IgE<20 IU/ml,补体C3 0.944 g/L,C4 0.192 g/L。ANA、A-dsDNA均阴性,类风湿因子阴性。
其他 外周血淋巴细胞亚群CD4 1052个/μl,CD8 785个/μl,CD3 1951个/μl,CD20 226个/μl,调节性T细胞46个/μl。CA125、CA199、CEA、PSA、AFP等肿瘤标志物均阴性。HBsAg、抗HCVIgG、抗HIV1/2,梅毒抗体检测均阴性。
辅助检查 双肾B超:左肾135 mm×59 mm× 65 mm,右肾124 mm×54 mm×60 mm,皮质厚度不清,双肾体积偏大,轮廓欠规则,包膜连续完整。胸片:正常。心电图:窦性心律。肝、胆B超:正常。甲状腺B超:甲状腺左叶腺瘤。眼底:A∶V=1∶2,动脉硬化I级,角膜、晶状体未见异常。上消化道气钡双重造影:正常。
肾活检病理
光镜 皮质和皮髓交界肾组织各1条,20个肾小球,1个球性硬化,2个节段硬化(图1A)。余正切球体积增大,直径(227.57±5.67)μm。肾小球足细胞胞质减少,毛细血管袢开放、僵硬,节段外周袢与囊壁粘连,系膜区节段轻~中度增宽,系膜基质增多为主,节段壁层上皮细胞增生,数处见球囊滴,囊壁增厚、分层。PASM-Masson:上皮侧较多嗜复红物沉积,见较多钉突形成(图1B、C)。肾小管间质慢性病变轻度,小灶性肾小管基膜增厚、萎缩,部分未萎缩小管基膜亦增厚,间质灶性纤维化,见多灶性泡沫细胞,部分呈簇状分布(图1D),并见少量单个核细胞浸润。小动脉节段透明变性。
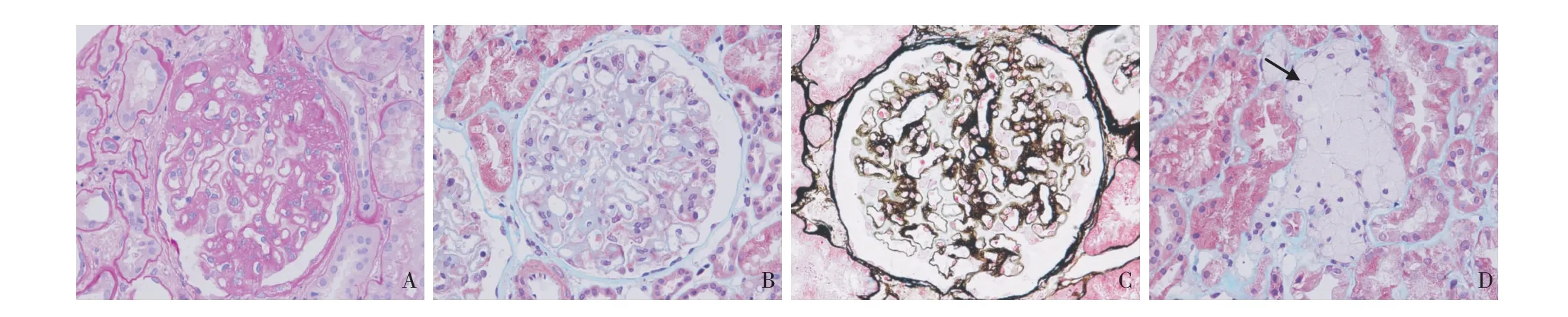
图1 A:肾小球系膜区轻度增宽,见节段硬化,袢与囊壁粘连(PAS,×400);B:肾小球毛细血管袢上皮侧弥漫嗜复红物沉积(Masson三色,×400);C:肾小球毛细血管袢僵硬,上皮侧钉突形成,一处袢与囊壁粘连(PASM-Masson,×400);D:肾皮质间质灶性呈簇状分布的泡沫细胞(↑)(M asson三色,×400)
免疫荧光 肾小球5个,荧光染色IgG++、IgA +、IgM+、C3++、C4+、C1q+,弥漫分布,呈颗粒状沉积于血管袢(图2)。肾组织IV型胶原:α3链染色肾小球基膜(GBM)强度明显减弱,肾小管基膜(TBM)阳性小管数量减少,α5链染色GBM、包囊壁(BC)、TBM均正常(图3)。
电镜 观察1个肾小球,基膜病变明显,基膜致密层不清晰,部分见分层撕裂,内外侧缘不规则,偶见呈典型“花篮样”改变(图4 A),节段分层、撕裂的基膜内见细颗粒样物质分布,直径约72~84 nm,有的呈中空状(图4 B),基膜上皮侧亦见类似颗粒样物质,及细小的电子致密物沉积,致密物周围见基膜样物质包绕,部分密度略减低(图4C)。肾小球足细胞足突广泛融合、增宽,较多微绒毛化。肾小球系膜区节段增宽,基膜样物质增多,未见电子致密物沉积。数处近端肾小管上皮细胞刷状缘及胞质成分脱落,有的小管胞浆质见脂性空泡。
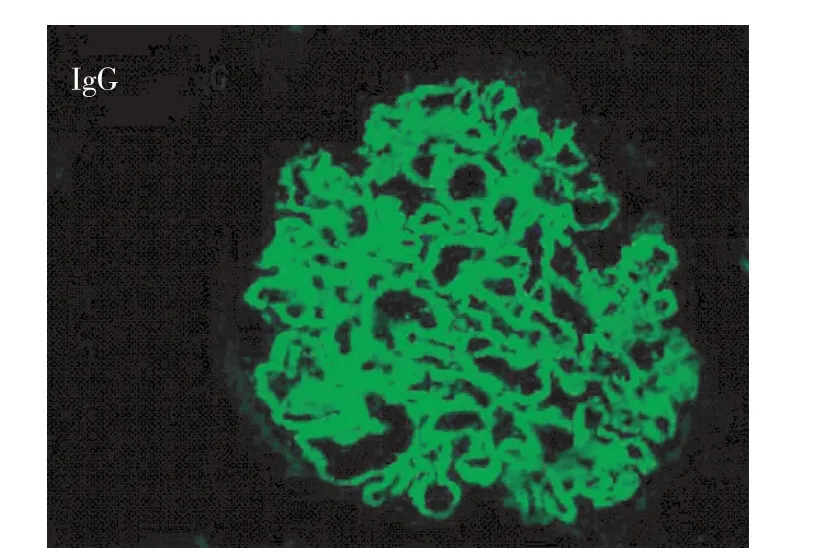
图2 A:IgG++沿肾小球毛细血管袢呈颗粒状弥漫分布(IF,×400)

图3 A:肾组织胶原IVα3链:肾小球GBM强度明显减弱(IF,×400);B:肾组织胶原IVα5链:肾小球GBM正常(IF,× 400);C:肾组织胶原IVα3链:TBM阳性小管数量减少(IF,×400);D:肾组织胶原IVα5链:TBM正常(IF,×400)

图4 A:肾小球基膜致密层消失,内外侧缘不规则,呈“花篮样”改变,足细胞足突广泛融合;B:肾小球基膜致密层分层、撕裂,基膜内见细颗粒样物质;C:肾小球基膜上皮侧电子致密物沉积,致密物周围见基膜样物质包绕,部分密度略减低(↑),足细胞足突广泛融合(EM)
小结:肾小球膜性病变III~IV期,但节段基膜分层、撕裂,结合肾组织Ⅳ型胶原α3链染色异常,考虑合并Alport综合征。
讨 论
该患者为中年男性,病程5年,临床表现为肾病综合征(NS),血压升高,无血尿,肾功能正常,外院肾活检诊断为膜性肾病(MN),曾予足量激素及多种免疫抑制剂治疗(包括环磷酰胺、雷公藤多苷等),病情始终无缓解,持续大量蛋白尿。为明确病变,患者入院后行重复肾活检,其肾组织光镜病理形态学较典型,表现为肾小球体积增大,毛细血管袢僵硬,上皮侧较多嗜复红物沉积,并见“钉突”形成,结合其免疫荧光IgG++弥漫血管袢沉积,符合肾小球膜性病变。
但除此之外,该患者病理上可见一些特殊之处:(1)肾间质可见多灶性泡沫细胞,部分呈簇状分布;(2)肾小球系膜区节段轻-中度增宽,见两处节段硬化,球囊粘连及囊壁增厚、节段分层;(3)间质存在与小球病变不相符合的灶性慢性化病变。一般来说,MN无肾小球固有细胞的增生,如果出现严重的系膜细胞增生、节段硬化,需要排除继发性病因的可能。因此我们首先对患者进行了MN继发因素的相关排查,包括乙型肝炎、自身抗体、肿瘤标志物等,但均未见异常。
其次,在患者光镜病理改变中尤其令人注意的是肾间质多灶性分布的泡沫细胞。肾间质泡沫细胞起源于单核细胞/巨噬细胞,可表达巨噬细胞的表型标志[1]。泡沫细胞不仅见于Alport综合征,也可出现于其他多种肾小球疾病中,特别是临床表现为长期NS的患者中,如特发性膜性肾病(IMN)、IgA肾病(IgAN)、局灶节段性肾小球硬化(FSGS)以及膜增生性肾小球肾炎等。不同肾脏疾病中间质泡沫细胞的发生率不同,其中以Alport综合征发生率最高(占64.8%),而FSGS、MN和IgAN患者间质泡沫细胞检出率依次为36.5%、21.4%和12.4%。但值得注意的是,不同疾病中肾间质泡沫细胞的分布方式是不同的:Alport综合征肾间质泡沫细胞有其独特之处,可单个散在分布,但多为灶性聚集,聚集的泡沫细胞相互挤压围成实心巢状,成簇状分布,量多时可连成片状;而MN、IgA肾病中泡沫细胞多为单个散在分布,灶性聚集较少见[2,3]。
本例患者病程长,持续大量蛋白尿不缓解,亦可导致间质泡沫细胞出现,但大量簇状泡沫细胞分布仍难以完全用MN解释。据此,建议患者行肾组织Ⅳ型胶原染色,结果示肾组织Ⅳ型胶原α3链染色异常,GBM强度明显减弱,TBM阳性小管数量减少;同时我们超微结构观察发现除上皮侧电子致密物及“钉突”外,肾小球基膜病变明显,基膜致密层不清晰,部分见分层撕裂,内外侧缘不规则,偶见典型“花篮样”改变。最终明确该患者诊断为:MN合并Alport综合征。
Alport综合征患者肾脏损害轻重不一,组织学改变可从轻微病变、系膜增生到局灶节段性肾小球硬化;肾小管间质病变如成簇状分布的泡沫细胞、肾小管萎缩及间质纤维化等。常规免疫荧光检查肾小球无免疫球蛋白和补体沉积,或仅见IgM和C3非特异性沉积[4,5]。因而依靠光镜病理和常规免疫病理,极易延误诊断;Alport综合征诊断必须依靠特殊免疫病理(包括肾组织Ⅳ型胶原、皮肤Ⅳ型胶原染色)和电镜检查,而且肾组织Ⅳ型胶原染色有助于判断遗传类型[6]。该例患者由于光镜下肾小球膜性病变表现典型,与其临床表现相符合,无明显血尿,无遗传性肾炎家族史及眼、耳的病变,若无肾组织Ⅳ型胶原及电镜检查的辅助,必将误诊为单纯的MN。这也提示在疾病诊断中必须要注重特殊免疫病理和电镜的作用。同时,在MN、IgAN、FSGS等光镜上表现有泡沫细胞时,应该注意观察泡沫细胞的分布形态,且思维要广阔,并不能满足于这些疾病本身也可以导致出现泡沫细胞这种想法,从而忽略了合并其他疾病的可能。该例患者即因为观察到多灶性簇状分布的泡沫细胞,通过进一步行肾组织Ⅳ型胶原及电镜检查,才作出了正确诊断。
临床上常可见两种性质不同的疾病同时出现在同一患者,如DN合并IgAN[7],MN合并DN[8]等。MN和Alport综合征均是常见肾脏疾病,根据南京军区南京总医院肾脏病研究所13 519例肾活检统计,MN及Alport综合征占肾活检的比例分别为6.79%和0.762%[9],但两者合并存在尚未见文献报道。本例患者经过光镜、免疫病理和电镜首次证实了MN及 Alport综合征可以合并存在,也提示疾病的诊断是综合性的,应该结合临床、实验室检查及病理组织学,注意临床与病理上的特殊性与不一致性,并注重特殊免疫病理和电镜在诊断中的重要作用。
该患者自肾活检后于我科随访两年余,先后给予激素+雷公藤多苷片、激素+他克莫司治疗,疗效不佳,持续大量蛋白尿(蛋白定量5.74 g/24h)、低蛋白血症(Alb 24.7 g/L)无缓解,肾功能尚在正常范围(SCr 79.56μmol/L)。根据南京肾脏病研究所对217例IMN患者的长期预后分析,发现肾活检后5年、10年及15年肾生存率分别为96.8%,93.0%和85.5%,起病初即有肾功能衰竭和在肾活检时病理上肾小管间质慢性化病变分级较高为MN患者进展为终末期肾功能衰竭的独立危险因素[10]。由于本例患者合并Alport综合征,且肾活检时即存在肾小管间质明显慢性化改变,免疫抑制剂治疗无效,其长期的肾脏存活尚待继续随访观察。
1 Franco M,Schmitt F,RejailiWA,etal.Renal interstitial foam cells are macrophages.Histopathology,1992,20(2):173-176.
2 Magil AB.Interstitial foam cells and oxidized lipop rotein in human glomerular disease.Mod Pathol,1999,12(1):33-40.
3 陈 丹,陈 燕,曾彩虹,等.肾小球疾病患者肾间质泡沫细胞的分布及其临床意义.肾脏病与透析肾移植杂志,2006,15(4):322 -328.
4 王 芳,丁 洁,俞礼霞,等.中国Alport综合征临床特征.临床儿科杂志,2003,21:601-604.
5 郭顺华,丁 洁,杨霁云.中国Alport综合征临床特点与研究现状.肾脏病与透析肾移植杂志,1999,8(01):71-74.
6 龚 伟,刘志红,周 虹,陈惠萍,等.Alport综合征IV型胶原分布特点及其与临床表型的联系,肾脏病与透析肾移植杂志,2003,12(03):201-206.
7 陈惠萍.糖尿病肾病合并IgA肾病.肾脏病与透析肾移植杂志,2009,18(6):584-587
8 金 波,刘志红,葛永纯,谢红浪等.肾活检患者中糖尿病肾病流行病学特点的变迁.肾脏病与透析肾移植杂志,2009,18(2):133 -139.
9 Li LS,Liu ZH.Epidemiologic data of renal diseases from a single unit in China:analysis based on 13519 renal biopsies.Kidney Int,2004,66:920-923.
10左 科,吴 燕,李世军.中国特发性膜性肾病患者长期预后及危险因素分析.肾脏病与透析肾移植杂志,2011,20(1):7-11.
M embranous nephropathy associated w ith A lport synd rome
HUANGQian,ZENGCai-hong,LIU Zhi-hong
Research Institute of Nephrology,Jinling Hospital,Nanjing University School of Medicine,Nanjing 210002,China
A 38-year-oldman presented with nephrotic syndrome and hypertension for five years.He had normal visual acuity and hearing,and denied hereditary nephropathy in his family.Renalbiopsy showed non-typicalmembranous nephropathy by the lightmicroscope and immunofluorescence.In addition,therewere severalunusual clusters of foam cells in the renal interstitial area.Type IV collagen staining showed that the expression of the alpha3 was faint in the GBM(glomerular basementmembrane)and normal in TBM(tubular basementmembrane).Electron microscope indicated the splitting of the lamina densa of GBM,and subepithelial electron dense deposits.The final diagnosiswas Alport syndrome associated withmembranous nephropathy.
Membranous nephropathy Alport syndrome renal biopsy
2011-02-21
(本文编辑 心 平 律 舟)
南京军区南京总医院全军肾脏病研究所(南京,210002)
猜你喜欢
包装工程(2022年5期)2022-03-21
中国土壤与肥料(2021年5期)2021-12-02
东方考古(2021年0期)2021-07-22
摄影之友(影像视觉)(2020年2期)2021-01-14
云南化工(2020年4期)2020-02-22
中国卫生标准管理(2015年15期)2016-01-15
医学美学美容·中旬刊(2015年2期)2015-10-21
中国塑料(2015年2期)2015-10-14
中国医疗美容(2015年4期)2015-04-27
食品科学(2013年13期)2013-03-11
