
HPLC法同时测定补肾壮骨颗粒中淫羊藿苷及柚皮苷含量
2011-05-26 01:13:00韩丽萍陈行愉邓伟民
中成药 2011年1期
韩丽萍, 陈行愉, 邓伟民
(广州军区广州总医院,广东广州 510010)
补肾壮骨颗粒由淫羊藿、骨碎补、鹿角胶等七味药材制成,具有补肾壮骨、健脾和胃、祛瘀止痛的作用,可用于各种原发性骨质疏松。其中淫羊藿和骨碎补为主药,淫羊藿中淫羊藿苷等黄酮类化合物为其主要活性成分,骨碎补其主要活性成分为柚皮苷等二氢黄酮类化合物。笔者曾对补肾壮骨颗粒中的淫羊藿苷进行测定[1],但对于一个复方制剂而言,仅对其中一种成分进行测定,还略显不足,为了更进一步地评价补肾壮骨颗粒的质量,本实验参考相关文献[2],采用HPLC法同时测定淫羊藿苷及柚皮苷含量。结果证明,该法操作简便,重现性好,且准确稳定,可为全面评价补肾壮骨颗粒的质量提供依据。
1 仪器与材料
Agilent 1100高效液相色谱仪:配有二元梯度泵、在线脱气机、自动进样器、柱温箱、DAD检测器、Chemstation色谱工作站(美国惠普公司),Sartorius BP211D分析天平(德国赛多利斯公司),KP-Q-1200超声波清洗机(广州科普超声电子技术有限公司)。
补肾壮骨颗粒(广州军区总医院制剂中心提供,批号分别为 081114,090402,090605,090921),淫羊藿苷、柚皮苷对照品(中国药品生物制品检定所,批号分别为110737-200415、110722-200610),95%乙醇、甲醇(分析纯)、乙腈(色谱纯,为SIGAM产品)。流动相用水为重蒸馏水。
2 方法与结果
2.1 色谱条件和系统适应性试验 色谱柱:Inertsil C18柱(4.6 mm ×250 mm,5 μm);流动相:A 相为水,B 相为乙腈,B相浓度梯度:0~8 min,体积分数为25%,8~9 min,体积分数为25% ~30%,9~22 min,体积分数为30%,检测波长285 nm;流速1 mL/min;柱温30℃;进样量10 μL。各峰的理论塔板数不低于3 000,与相邻的峰的分离度均大于1.5。在上述色谱条件下,对照品、样品和阴性样品的色谱图见图1。
2.2 线性关系考察 分别取淫羊藿苷及柚皮苷对照品适量,精密称定,置10 mL量瓶中,用甲醇溶解并稀释至刻度,制成1 mL约含淫羊藿苷0.6 mg,柚皮苷0.4 mg的混合对照品贮备溶液。精密吸取对照品贮备溶液适量,加甲醇分别稀释成每毫升含柚皮苷约为 0.4、0.08、0.04、0.016、0.008、0.004 mg,含淫羊藿苷约为 0.6、0.12、0.06、0.024、0.012、0.006 mg的对照品系列溶液,精密吸取10 μL,注入液相色谱仪,以峰面积对质量浓度进行线性回归,得各组分的线性范围和回归方程,淫羊藿苷在进样量0.060 6~6.6 μg范围内线性关系良好,Y=1 053.6X+9.400 7,r=0.999 9;柚皮苷在进样量0.087 4~8.74 μg范围内线性关系良好,回归方程:Y=1 538.8X+27.511,r=0.999 9。
淫羊藿苷和柚皮苷的最低检出限分别为20.2 ng和29.1 ng,定量限分别为52.5 ng和75.7 ng。
2.3 供试品溶液制备方法考察 精密称取补肾壮骨颗粒约2 g,置圆底烧瓶中,精密加入甲醇50 mL,精密称定,考察下列提取方法:①浸泡2 h,超声提取30 min;②回流提取30 min;③浸泡2 h,超声提取30 min,继续回流提取30 min。上述3种提取液放冷至室温,以甲醇补足损失,滤过。精密吸取续滤液25 mL至蒸发皿中,水浴蒸干,残渣用甲醇溶解,置10 mL量瓶中,用甲醇稀释至刻度,摇匀,0.22 μm微孔滤膜滤过,作为供试品溶液。
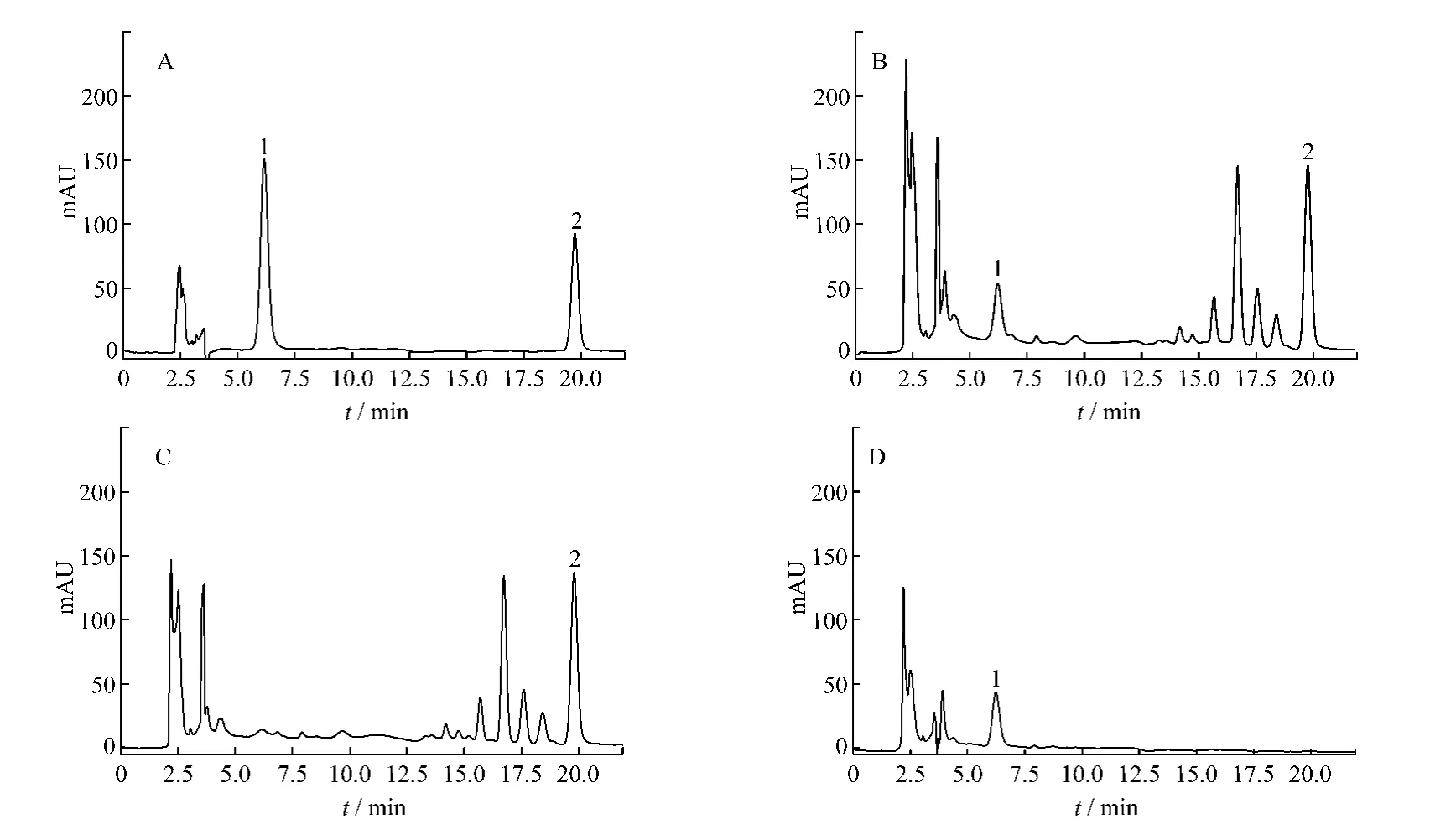
图1 补肾壮骨颗粒HPLC色谱图
取上述3种供试品溶液,按照2.1项下方法,测定淫羊藿苷和柚皮苷含量,结果表明,回流提取与超声提取对淫羊藿苷影响较小,而柚皮苷用回流提取优于超声提取,但单纯超声或回流提取,两种成分均不能提取完全,因此确定采用超声加回流的方法进行提取,见表1。

表1 不同制备方法对含量的影响(n=3)
2.4 精密度试验 按照2.3项下第三法制备供试品溶液,按照2.1项色谱条件下进样10 μL,重复6次,以淫羊藿苷和柚皮苷峰面积计算RSD,其结果分别为0.56%、0.58%,说明仪器精密度良好。
2.5 重复性试验 取同一批补肾壮骨颗粒样品(批号:090921),按照2.4项下第三法平行制备6份供试品溶液,按照2.1项色谱条件下进样10 μL,计算淫羊藿苷和柚皮苷含量的RSD分别为1.17%、1.39%,说明该法重复性较好。
2.6 稳定性试验 取同一供试品,按照2.1项色谱条件分别在0、1、2、4、6、8、24 h 进样10 μL,测定峰面积,计算淫羊藿苷和柚皮苷峰面积的RSD分别为0.88%、1.15%。表明供试品溶液在24 h内稳定性良好,可满足测定要求。
2.7 加样回收率试验 取已知淫羊藿苷、柚皮苷含量的补肾壮骨颗粒(批号:090921)6份各约1 g,精密称定,置圆底烧瓶中,精密加入淫羊藿苷、柚皮苷对照品甲醇溶液适量,按2.4项下方法制备供试品溶液,按2.1项下色谱条件测定含量,求得淫羊藿苷和柚皮苷的回收率,结果见表2。
2.8 样品含量测定 取大生产的4批样品,按2.4项下方法制备供试品溶液,按2.1项下色谱条件,分别将混合对照品和供试品溶液进样10 μL,记录淫羊藿苷和柚皮苷的色谱峰面积,外标法计算含量,结果见表3。

表2 加样回收率试验结果(n=6)

表3样品测定结果(n=3)
3 讨论
由于柚皮苷和淫羊藿苷性质差异较大,为了能使两组分能按各自适宜的容量因子k达到良好的分离目的,以不同比例的乙腈-水为流动相对色谱条件进行考察。结果表明,当流动相为乙腈-水的起始浓度为25:75时,各组份能达到较好的分离。采用乙腈-水单一比例测定淫羊藿苷约10 min[1],但柚皮苷不能很好分离。骨碎补药材中新北美圣草苷和柚皮苷的含量同时测定用时为20 min[3],本方法2组分同时测定用时约20 min,缩短了多组份检测的分析时间。进一步的对比研究发现,梯度洗脱可以消除杂质对被测成分的干扰,避免因使用聚酰胺进行净化处理[1]导致被测成分的损失,大大节省了样品制备的时间及溶剂,适于全面评价补肾壮骨颗粒的质量。
淫羊藿苷的最大吸收波长为270 nm,柚皮苷的最大吸收波长为285 nm,本实验考察了在波长为270及285 nm条件下的吸收图谱,发现在285 nm单一波长下,淫羊藿苷及柚皮苷同时可以得到较好的色谱峰。也可用双通道检测,在270 nm波长下检测淫羊藿苷,在285 nm下波长检测柚皮苷。
本次实验对不同批次补肾壮骨颗粒含量进行测定,结果显示,不同批次样品之间淫羊藿苷含量差异较大,与笔者之前的报道[1]不相符,主要原因是报道中的3批样品均使用同一批次药材制备,而本次实验的样品使用不同批次药材制备,但市面上淫羊藿药材的质量参差不齐,笔者测定不同批次的淫羊藿药材,淫羊藿苷含量差异可达到10倍以上,因此控制原药材质量,是保证中药制剂质量的根本措施。
[1]韩丽萍,梁 达,刘志刚,等.补肾壮骨颗粒质量标准的研究[J]. 解放军药学学报,2009,25(2):145-147.
[2]李 超,葛 洁,鹿秀梅,等.RP-HPLC法测定骨疏丹颗粒中5种有效成分的含量[J].沈阳药科大学学报,2008(9):728-731.
[3]李遇伯,孟繁浩,李发美,等.HPLC同时测定骨碎补药材中新北美圣草苷和柚皮苷的含量[J].沈阳药科大学学报,2006,23(6):808-810.
猜你喜欢
食品与发酵工业(2023年21期)2023-11-26 07:50:24
江西中医药(2022年8期)2022-08-22 02:01:26
口腔医学(2021年10期)2021-12-02 02:07:44
中成药(2018年12期)2018-12-29 12:26:00
中国中医药现代远程教育(2018年22期)2018-02-09 02:12:04
湖南林业科技(2017年6期)2018-01-30 03:48:06
中成药(2017年4期)2017-05-17 06:09:49
中国医药科学(2016年9期)2016-07-25 23:16:55
饲料博览(2016年7期)2016-04-05 14:20:34
陕西中医(2015年11期)2015-03-22 04:29:17
