
变形链球菌gcp基因突变菌株的构建及鉴定*
2011-05-24 07:53闫文娟杨德鸿吴补领
中华老年口腔医学杂志 2011年6期
闫文娟 杨德鸿 吴补领
牙菌斑生物膜是龋病的始动因子,变形链球菌是公认的主要致齲菌,变形链球菌在牙齿表面黏附、聚集并形成菌斑生物膜是其致龋的关键因素。环鸟苷二磷酸(3',5'-Cyclic diguanylic acid;c-di-GMP)是近年来发现的细菌第二信使,它的作用涉及生物膜形成、毒力因子的表达等多个方面,是细菌生存和代谢的关键性调节因子之一[1,2]。在前期的研究中我们证实变形链球菌内部存在c-di-GMP信号通路,可影响变形链球菌的生物膜形成和在牙齿表面的黏附等,但其作用机制未明,基因重组技术为变形链球菌致龋毒力基因功能的研究提供了一个有力的工具[3]。本研究拟构建变形链球菌中c-di-GMP相关蛋白基因gcp(AEO14133)基因突变株,为下一步的变形链球菌c-di-GMP功能及多种防龋机制的研究奠定基础。
1.材料与方法
1.1 菌株和质粒 S.mutans UA159(四川大学华西口腔医院);E.coli Top 10、pMD-19T质粒(大连宝生物工程有限公司);PVA8912自杀载体(College of Newcastle,England),全长约 2.9 kb,含有多克隆位点。
1.2 主要试剂 LA Taq DNA聚合酶、T4DNA连接酶和限制性内切酶(大连宝生物工程有限公司);凝胶回收试剂盒(Clontech公司);质粒小量提取试剂盒(Omega公司);细菌基因组DNA提取试剂盒、DNA Marker(北京天为时代公司);LB培养基、BHI培养基(Oxoid,England,CM225),氨苄青霉素(Amp)浓度100mg/mL,红霉素分别为 400-500 mg/L(E.coli)和 20 mg/L(S.mutans)。
1.3 变形链球菌UA159的DNA提取 用磷酸盐缓冲液(PBS)复苏变形链球菌UA159冻干粉,于BHI培养基划线复苏培养,经革兰氏染色和生化鉴定后,在厌氧箱内(N2:85%、CO2:5%、H2:10%)37℃厌氧培养48h,挑取单克隆接种于新鲜配制的BHI培养液中,培养24h,取3ml菌液,按照细菌基因组DNA提取试剂盒的说明提取DNA,凝胶电泳鉴定后-20℃保存备用。
1.4 PCR扩增gcp基因内部片段 通过Genebank查询gcp基因(ID:AEO14133)上下游同源碱基序列,设计一对PCR引物,上游引物为:5’-TTACGCATGCATGATTTTGTTTGGAATT TTGGC-3’(下划线代表Sph I酶切位点),下游引物为:5’-TAGCGAATTCTTTCACGGATAACA GCTTGGTC-3’(下划线代表EcoR I酶切位点)。以细菌DNA片段为模板,配制50μl的反应体系,通过PCR扩增873bp内部序列,对应于Gcp蛋白的19-310氨基酸序列。PCR扩增结束后,对PCR产物进行凝胶电泳检测,并用DNA凝胶回收试剂盒纯化回收。
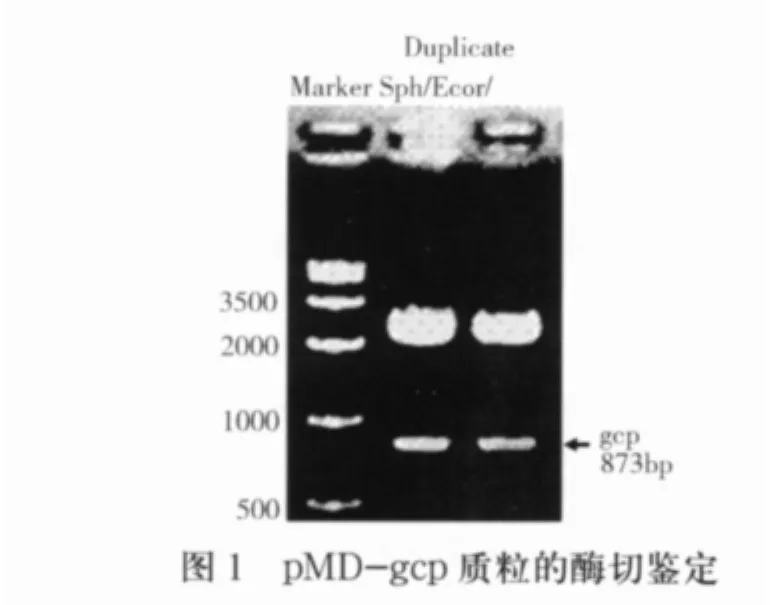
1.5 PCR产物的重组及克隆 将纯化后的PCR扩增产物和pMD-19T载体连接,16℃孵育16h;连接产物转化至E.coli Top10感受态细胞中。将转化细菌涂布于含Amp的LB固体培养基平板上,37℃培养过夜。挑取单克隆接种于含Amp的LB培养液中37℃振摇过夜培养,用质粒小量提取试剂盒提取质粒DNA,用SphI/EcoRI酶切鉴定。鉴定正确的克隆命名为pMD-gcp。
1.6 打靶载体pVA8912-gcp的构建及鉴定将pVA8912质粒转化至E.coli Top10感受态细胞中,进行扩增,用质粒小量提取试剂盒纯化获得的质粒DNA以及pMD-gcp质粒,用SphI/EcoRI双酶切,DNA凝胶电泳检测并切胶纯化。取gcp内部序列片段和线性化pVA8912片段按照摩尔比3∶1的比例,用T4 DNA连接酶于16℃连接过夜,转化E.coli Top10感受态细胞后,在含红霉素的LB培养板上筛选阳性克隆,用质粒小量提取试剂盒提取质粒,获得pVA8912-gcp。
1.7 变形链球菌感受态细胞的制备及转化 挑取变形链球菌UA159单克隆接种于BHI液体培养基培养18 h,取100 μL变形链球菌液接种入5mL的BHIS(含5%马血清),37℃厌氧培养18h。从上述菌液中取出100ul加入到37℃预热的BHIS中,培养3h,加入10μg/mL的pVA8912-gcp质粒,轻轻晃动后37℃孵育30min,加入1ml 37℃预热的BHIS继续孵育90 min,涂布于含红霉素的BHI平板上,37℃厌氧培养48 h。
1.8 变形链球菌UA159gcp基因突变菌株的筛选及鉴定 选取位于BHI抗性平板上的阳性克隆菌落数个,分别接种于含红霉素的BHI液体培养基中,经鉴定证实为变形链球菌。提取突变菌株基因组DNA,采用PCR法进行突变菌株的鉴定。依据变形链球菌UA159 gcp序列设计引物,扩增包含gcp基因上游119bp的片段,引物序列如下(5’-3’):P1:5’-TCTATCTTGAAGGAAAGAATAGAAGTC-3’;P2:5’-GCGACTCCGTGCAGATATACAA AC-3’。利用引物P1和P2自变形链球菌UA159 gcp功能丧失菌株中扩增目的片段,PCR产物回收后用PstI酶切扩增片段,电泳观察。
2.结果
2.1 gcp基因内部片段的克隆 通过PCR扩增得到gcp基因编码区(57 bp-930 bp)的内部序列,全长为873 bp,连接pMD-19T载体后,经SphI/EcoRI酶切鉴定结果见图1。通过以上鉴定初步可以认为gcp基因,经过序列测定,结果正确。
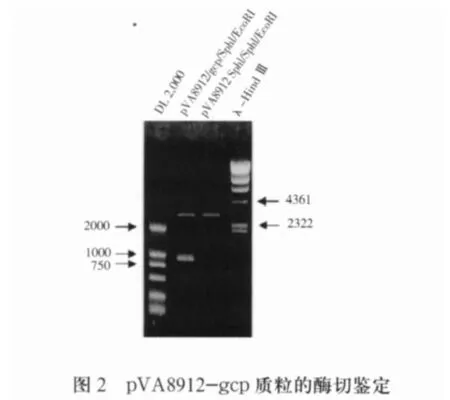
2.2 打靶载体pVA8912-gcp的鉴定 自杀载体pVA8912来源于质粒pVA891,带有红霉素抗性基因,能够在大肠杆菌中复制,但不能在链球菌中复制。采用SphI/EcoRI双酶切自pMD-gcp质粒中切出包含gcp基因内部序列的片段,定向插入同样酶切的自杀载体pVA8912中,构建gcp基因打靶载体pVA8912-gcp,873bp的gcp基因内部片段克隆入SphI/EcoRI双酶切后的pVA8912质粒中,构建gcp基因打靶载体pVA8912-gcp。转化S.mutans后,载体骨架通过同源重组插入S.mutans基因组中gcp基因内部,使gcp基因失活,酶切鉴定结果见图2。
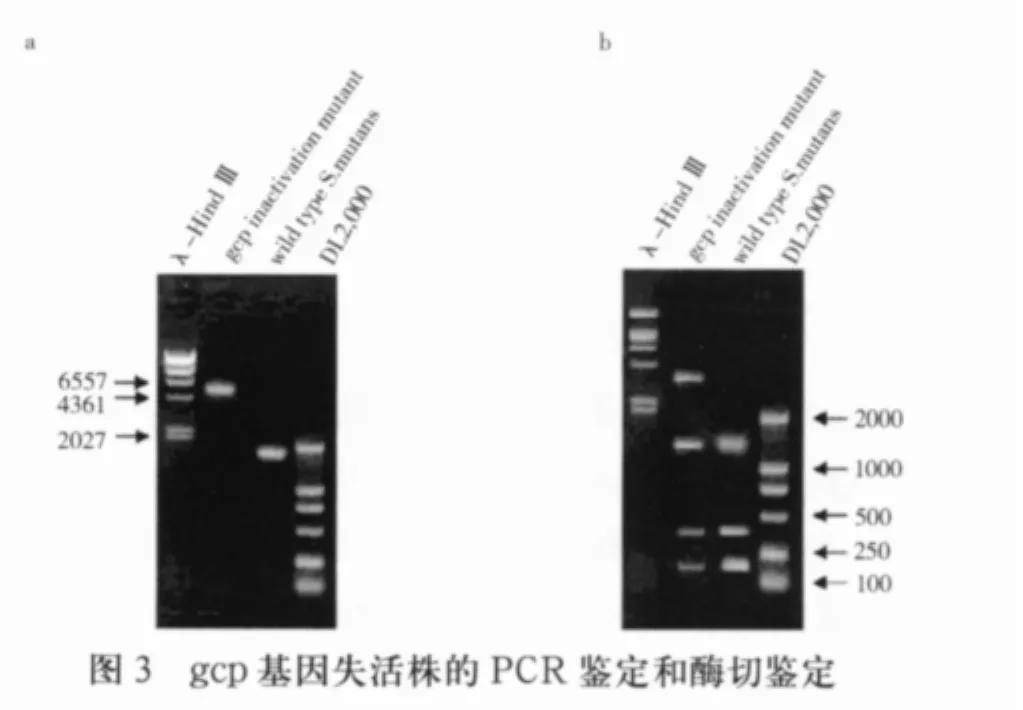
2.3 gcp基因失活突变菌株的鉴定 以变形链球菌UA159基因组DNA为模板扩增出1913bp片段,而以gcp突变菌株基因组DNA为模板扩增得到5742 bp片段,电泳结果如图3a所示,与预期结果相符,证实了突变菌株中的gcp基因被成功替代。为排除PCR结果假阳性的可能,将PCR产物回收,用PstI单酶切,电泳结果显示,突变型菌的PCR产物可以切出四条带,比野生型菌多出一条约3.8 kb的条带,与预期结果相符(图3b)。
3.讨论
基因缺失是目前研究基因功能最常用的技术之一,该技术将外源性DNA分子引入受体细胞,使之获得新的遗传性状,这一过程需要质粒来介导供体DNA。自杀质粒应具备以下条件:①自杀质粒在受体菌中不能复制;②自杀质粒必须带有一个在整合到染色体内以后可供选择的抗性标记;③自杀质粒带有易于克隆的多克隆位点。在变形链球菌的研究中最常用的自杀质粒是R质粒的衍生质粒。该质粒在进行复制时需要一种特殊的蛋白质,但是大多数细菌不产生这种蛋白质。因此,当自杀质粒进入受体菌时,只有整合到受体菌染色体中才能进行复制。在基因功能研究中根据自杀质粒的特点,将目的基因片断克隆入自杀质粒中,利用同源重组原理[4],定位自杀质粒的整合位点,构建特定基因缺失的菌株,从而研究该基因的功能[5]。本实验为深入研究gcp基因与变形链球菌致龋性之间的关系,需利用同源重组原理构建gcp基因功能丧失菌株[6],因此本实验工作的重要内容之一就是构建符合同源重组条件的合格质粒载体。
本实验中所采用的自杀载体pVA8912是链球菌自杀载体之一,含有红霉素抗性基因和多克隆位点,可以作为筛选标志[7]。同时自杀载体pVA8912只具有E.coli的复制起始区序列但是缺乏链球菌的复制起始区序列,因此能够在E.coli中复制而不能够在链球菌中复制。在转化链球菌后,可以通过单次交换同源重组的方式整合到细菌基因组中。这种转化的效率要比采用线性DNA转化的二次交换同源重组的效率高104倍左右[8]。同时,实验中还应用同源基因DNA片段构建载体,提高了同源重组的发生频率[9]。利用自杀载体转换链球菌时需要选择同源序列的长度和位置。如果同源序列过短,可能会造成同源重组的机率过低,而较难获得中靶克隆基因;如果同源序列过长,又可能会造成打靶载体过大,操作不方便。同时,同源重组后会产生两个不完整的gcp结构基因构成的串联重复序列,如果同源序列过长,外源基因插入后的gcp基因仍可能会有截短了的功能区蛋白表达,从而不能完全失活该基因。
本实验中同源DNA臂的长度在0.35 kb到2.15 kb之间时,随着DNA同源片段长度的增加,同源重组频率呈指数增加[10,11]。转化完成后,形态学观察及生化检测发现转化菌株形态及生化特性与变形链球菌标准菌株基本一致。以转化菌株的基因组DNA为模版进行PCR,结果显示这一区域的片段长度由原来的约2.5kb增加至约6.2kb,说明gcp基因内插入了其它的DNA片段。对PCR产物进行进一步酶切分析,得到的酶切片段的大小和数量均与预期相符,从核酸水平上证明了打靶载体成功的插入到gcp基因中,为进一步的gcp功能研究提供了有价值的细菌模型。
本实验根据同源重组原理,利用自杀载体PVA8912成功构建了gcp基因失活菌株,为以后研究gcp基因在变形链球菌中的致龋机制及功能奠定基础。
[1]SchirmerT and JenalU.Structuraland mechanistic determinants of c-di-GMP signaling[J].Nat Rev Microbiol,2009,7(10):724-735
[2]Jonas K,Melefors O,and Romling U.Regulation of c-di-GMP metabolism in biofilms[J].Future Microbiol,2009,4:341-358
[3]Yan W,Qu T,Zhao H et al.The effect of c-di-GMP(3'-5'-cyclic diguanylic acid)on the biofilm formation and adherence of Streptococcus mutans[J].Microbiol Res,2010,165(2):87-96
[4]段 劲,刘筱娣,郭丽宏.变形链球菌UA159磷酸蔗糖变位酶基因功能丧失菌株的构建 [J].实用口腔医学杂志,2008,24(1):69-73
[5]TaoL,LeBlancDJ,FerrettiJJ.Novelstreptococcal-integration shuttle ectors for gene cloning and inactivation [J].Gene,1992,120(1):105-110
[6]Merritt J,Tsang P,Zheng L et al.Construction of a counterselection-based in-frame deletion system for genetic studies of Streptococcus mutans[J].Oral Microbiol Immunol,2007,22(2):95-102
[7]Xie Z,Okinaga T,Qi F,et al.Cloning-independent and counterselectable markerless mutagenesis system in Streptococcus mutans[J].Appl Environ Microbiol,2011,77(22):8025-8033
[8]Tao L.Streptococcal integration vectors for gene inactivation and cloning[J].Methods Cell Sci,1998,20:59-64
[9]Bennett PM.Plasmid encoded antibiotic resistance:acquisition and transfer of antibiotic resistance genes in bacteria[J].Br J Pharmacol,2008,153(1):S347-357
[10]Indranil Biswas, Alexandra Gruss, Dusko Ehrlich.High-efficiency gene inactivation and replacement system for gram-positive bacteria [J].Journel of bacteriology,1993,175(11):3628-3635
[11]Rong R,Slupska MM,Chiang JH et al.Engineering large fragment insertions into the chromosome of Escherichia coli[J].Gene,2004,336(1):73-80
猜你喜欢
四川生理科学杂志(2022年4期)2022-12-06
环球时报(2022-09-20)2022-09-20
汉字汉语研究(2021年2期)2021-08-30
皮肤病与性病(2021年3期)2021-07-30
今日农业(2020年24期)2020-12-15
汉字汉语研究(2019年2期)2019-08-27
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
中华老年口腔医学杂志(2016年6期)2017-01-15
河北书画研究(2016年3期)2016-04-28
中国医药生物技术(2015年4期)2015-12-26
