
慢性阻塞性肺疾病患者支气管黏膜活检标本中 HO-1和 iNOS的表达
2011-04-01 10:45李巧荣吴大玮山东大学齐鲁医院加强医疗科山东济南500
中国老年学杂志 2011年14期
李巧荣 吴大玮 杨 蕾 韩 进 (山东大学齐鲁医院加强医疗科,山东 济南 500)
氧化应激反应在慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)的发病过程中起到重要作用〔1〕。炎症和氧化应激可以刺激细胞内血红素氧化酶-1(HO-1)和诱生型一氧化氮合酶(iNOS)的表达〔2〕。HO-1的降解产物具有抗氧化活性,HO-1还可直接上调细胞外过氧化物歧化酶(ec-SOD)表达,从而对肺脏有保护功能〔3,4〕;而 iNOS可产生过高的NO,使细胞的氧化还原电位转向更强的氧化状态;同时,NO与烟草中的超氧阴离子反应生成过氧亚硝酸离子和过氧亚硝酸盐,前者为强氧化剂,后者有细胞毒作用〔5〕。目前对于不同严重程度、稳定期或急性加重期 COPD患者肺组织 HO-1表达水平升高或降低仍有争议〔6,7〕,研究方法也主要是通过诱导痰、支气管肺泡灌洗液、手术切除标本进行〔6~8〕,对于活体支气管黏膜标本进行研究的报道少见。因此,本文经支气管镜钳取COPD患者支气管黏膜,观察 HO-1和 iNOS的表达变化。
1 材料与方法
1.1 病例选择 在山东大学齐鲁医院门诊或病房因肺部占位病变行支气管镜检查的患者 28例,根据是否吸烟和肺功能检查结果将其分为 3组:(1)吸烟伴 COPD组 9例,(2)吸烟肺功能正常组 9例(FEV1≥70%预计值);(3)不吸烟肺功能正常组10例。各组研究对象临床指标见表 1。吸烟两组之间的吸烟指数及三组患者在 FEV 1、年龄和性别方面的比较均无统计学差异。所有入选的 COPD患者均为稳定期,符合中华医学会呼吸病学分会慢性阻塞性肺疾病学组制定的《慢性阻塞性肺疾病诊治指南》中的诊断标准〔9〕。所有患者在术前 3~5 d内均进行了CT检查、常规肺功能检查和支气管舒张试验,排除肺间质纤维化、支气管扩张等其他影响肺功能的疾病。电子支气管镜黏膜活检时间均在上午 8:00~10:00。
1.2 黏膜取材 术前 10~12 h禁食,术前 15 min雾化吸入2%利多卡因行气道麻醉。仰卧位,给予鼻塞吸氧和心电监护,支气管镜通过鼻腔进入气道,用活检钳取肺肿块对侧(健侧)肺上/下叶(中间)、中/下叶支气管间脊黏膜。活检组织立刻放入多聚甲醛中固定、梯度酒精脱水、透明后石蜡包埋,做 2μm厚切片备免疫组化用。
1.3 免疫组化染色 用抗生素蛋白-生物素-过氧化物酶(ABC)法行细胞因子免疫组化染色,HO-1和iNOS为兔抗人多克隆抗体(购自北京中山生物技术有限公司),HO-1为即用型、iNOS为浓缩型(所用浓度为 1∶200)。将石蜡切片烘烤脱蜡至水,0.01mmol/L枸橼酸钠高压 10 min,进行抗原修复,室温冷却后 3%过氧化氢浸泡 10 min,10%山羊血清封闭 15 min,然后加一抗 4℃过夜,加二抗,37℃孵育 30 min,DAB显色,苏木素复染,脱水透明,树胶封片。HO-1和 iNOS均在胞质中表达,在显微镜 400倍下用全自动图像分析系统对阳性染色进行分析,测定两者的平均吸光度值,随机测定 4个高倍视野后计算平均值,作为该切片的代表值。
1.4 统计学分析 用SPSS11.5统计软件进行数据处理,所有数据以±s表示。各组免疫组化结果之间的比较采用两样本均数 t检验。HO-1和 iNOS及与 FEV1的相关性采用 Pearson相关分析。
2 结 果
2.1 免疫组化染色结果
2.1.1 HO-1的表达 HO-1在吸烟伴 COPD组患者气管黏膜的上皮细胞、巨噬细胞均呈强阳性表达,吸烟肺功能正常组表达增强,不吸烟肺功能正常组弱表达。见图 1。HO-1在吸烟伴COPD组(0.156±0.036)和吸烟肺功能正常组(0.147±0.049)的表达均明显高于不吸烟肺功能正常组(0.094±0.043)(P<0.01),吸烟两组无统计学差异(P>0.05)。
表1 研究对象临床资料(±s)
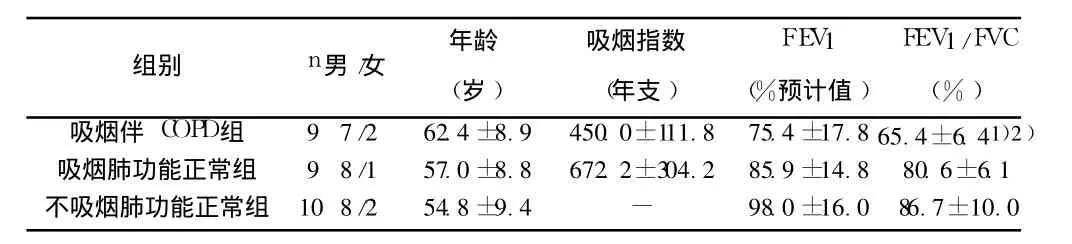
表1 研究对象临床资料(±s)
与吸烟肺功能正常组比较:1)P<0.01,与不吸烟肺功能正常组比较:2)P<0.01
吸烟伴 COPD组 9 7/2 62.4±8.9 450.0±111.8 75.4±17.8 65.4±6.41)2)吸烟肺功能正常组 9 8/1 57.0±8.8 672.2±304.2 85.9±14.8 80.6±6.1不吸烟肺功能正常组 10 8/2 54.8±9.4 - 98.0±16.0 86.7±10.0

图1 各组 HO-1和 iNOS的免疫组化染色(DAB,×400)
2.1.2 iNOS的表达 iNOS在吸烟伴 COPD组气管黏膜上皮细胞和巨噬细胞呈强阳性表达,在吸烟肺功能正常组呈阳性表达,在不吸烟肺功能正常组呈弱阳性表达。见图 1。iNOS在吸烟伴 COPD组的表达(0.188±0.030)明显高于不吸烟肺功能正常组(0.159±0.034,P<0.01),吸烟肺功能正常组(0.176±0.053)虽然高于不吸烟肺功能正常组(0.159±0.034),但两者之间无统计学差异(P>0.05)。
2.2 HO-1与 iNOS的表达与肺功能的关系 气管黏膜 HO-1与 iNOS的表达无明显的相关性(r=0.046,P>0.05),HO-1在气管黏膜中平均吸光度值与FEV 1呈负相关(r=-0.558,P<0.05)。见图 2。
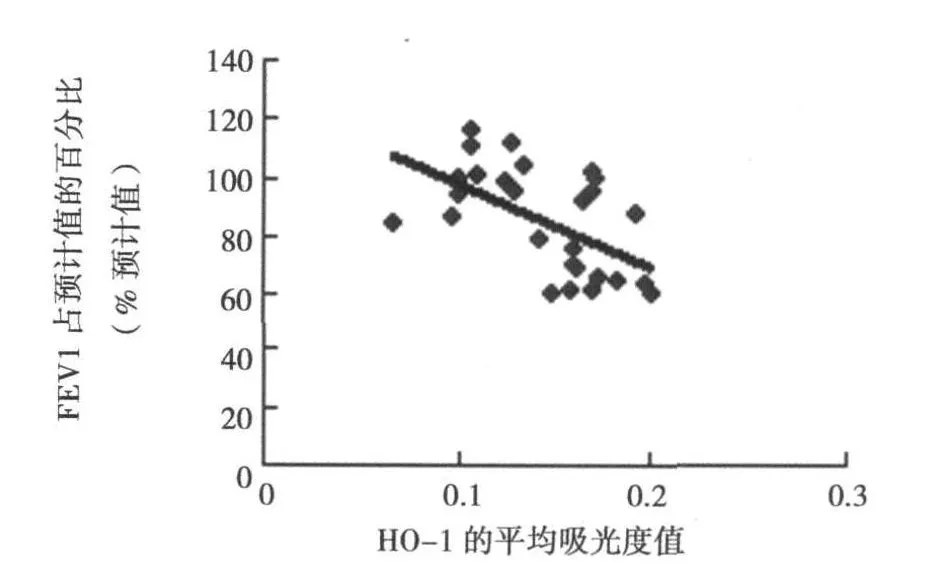
图2 HO-1平均吸光度值与 FEV 1的相关性
3 讨 论
HO是一种可诱导的抗氧化酶,目前发现有 3种同工酶,包括诱导型 HO-1、结构型 HO-2和 HO-3〔10〕。 目前发现 HO-2的表达和活性不受氧化应激等诱导,HO-3缺乏活性,其生物学特性和功能尚不清楚,而 HO-1是迄今为止发现的生物体内最容易被诱导产生的抗氧化酶。HO-1可以催化含铁血红素产生胆红素、铁蛋白和一氧化碳。研究发现胆红素是一种有效的抗氧化剂,体外实验证实胆红素能清除氧自由基,一氧化碳对炎症细胞或外源性氧化剂产生的氧自由基导致的肺损伤有保护作用〔11〕。
近年的研究还进一步发现,HO-1对于 COPD发病的多个病理和病理生理环节均有保护作用。Shinohara〔12〕的动物实验证明,腺病毒转基因小鼠肺组织 HO-1高表达可减低肺泡灌洗液中性粒细胞、TNF-α、IL-6和胶质细胞源性趋化因子水平,升高抗炎性因子 Il-10水平,并减轻弹性蛋白酶导致的肺气肿程度;Lee等〔13〕在人气道平滑肌细胞体外培养中加入 HO-1诱导剂钴原卟啉(cobalt protoporphyrin),或用携带人 HO-1基因的重组腺病毒感染平滑肌细胞,高表达的 HO-1通过下调氧化应激反应、黏附分子和 IL-6减轻了 TNF-α介导的气道炎症,抑制了 TNF-α诱导的 NF-κB活性,以及 TNFR1/c-Src/p47phox复合物的形成;HO-1可诱导牛肺动脉(BPA)细胞外过氧化物酶(ecSOD)表达增加,从而减轻低氧性肺血管收缩(HPV)〔3〕;Almolki等〔14〕报道 HO-1可抑制香烟暴露导致的气道黏液高分泌。
尽管 HO-1作为炎症和氧化应激的可诱导酶,但是,关于临床COPD患者肺和气道组织HO-1表达水平是升高抑或是降低报道结果不一。大部分研究〔6,8〕认为在 COPD患者肺和气道组织中HO-1表达增加,但也有部分作者报道 HO-1在COPD手术切除标本中表达降低〔7〕。本文通过支气管活检标本的研究表明,吸烟伴COPD组和吸烟肺功能正常组气管黏膜中 HO-1的表达均较不吸烟肺功能正常组增高,表明吸烟是诱导气道黏膜组织高表达 HO-1的重要原因。
以往少有肺功能与HO-1表达水平之间关系的报道。本研究发现,支气管黏膜中HO-1的表达水平与 FEV1呈负相关,提示随着 COPD程度加重,气道黏膜 HO-1表达增加。Hogg等〔15〕曾报道,随着COPD患者 GOLD分级增加,气道黏膜浸润的巨噬细胞和嗜中性粒细胞数也相应增加。根据生理学规律,随着炎症程度加重,其诱导 HO-1表达的水平也可能随之增高;但是,HO-1的抗氧化功能并不足以对抗炎症加重或长期病程对气道的氧化损伤作用〔6〕。曾有报道,在严重 COPD病人肺泡巨噬细胞内 HO-1表达减少,认为存在 HO-1基因多态性现象,使部分患者对氧化损伤的防御机制降低〔16〕。
iNOS主要由巨噬细胞产生,可以催化L-精氨酸生成一氧化氮(NO),过高的 NO可以造成组织细胞的氧化损伤〔5,17〕,活化基质金属蛋白酶,促进肺气肿的产生〔18〕。目前研究已经证明,COPD患者诱导痰液iNOS的表达升高,呼出气中的 NO含量增加〔19〕。本研究也表明,COPD病人气管黏膜中 iNOS的表达增强。但是,吸烟肺功能正常组 iNOS的表达尽管较不吸烟肺功能正常组增高,但无统计学差异。这在一定程度上提示,iNOS在COPD病人黏膜内表达增高可能不单纯是由于吸烟引起的,吸烟导致肺脏的炎症程度与黏膜内 iNOS表达之间的关系尚需要进一步的研究。
氧化/抗氧化机制的相互制约是一个复杂的过程。氧化应激可活化上皮细胞和炎症细胞内的转录因子,使编码 HO-1和iNOS的基因表达上调〔18〕。HO-1可以通过降解血红素下调 iNOS的表达,而 NO有稳定 HO-1信使核糖核酸的作用,从而可以上调 HO-1的表达〔20〕。本实验研究结果显示 COPD患者支气管黏膜中 HO-1和iNOS表达无明显相关性,不能确定这是否与本试验对象多为轻、中度 COPD病例有关,也不清楚重度COPD病例支气管黏膜中两者之间的相关关系。
本研究结果表明,气道黏膜 HO-1表达与气流受限程度正相关,这在一定程度上提示炎症和氧化应激诱导的抗氧化保护机制是不充分的,进一步探索可用于临床的强效HO-1活化剂可能是 COPD治疗研究的一个潜在方向。
1 Global Strategy for Diagnosis,Management,and Prevention of COPD〔J〕.Updated December,2009.
2 Wedzicha JA.Exacerbations:etiology and pathophysiologic mechanisms〔J〕.Chest,2002;121(suppl):136S-41S.
3 Ahmad M,Zhao XM,Kelly MR,et al.Heme oxygenase-1 induction modulates hypoxic pulmonary vasoconstriction through upregulation of ecSOD〔J〕.Am J Physiol Heart Circ Physiol,2009;297(4):H1453-61.
4 Morse D,Choi AM.Heme oxygenase-1:the“emerging molecule” has arrived〔J〕.Am J Respir Cell Mol Biol,2002;27:8-16.
5 Kroncke KD,Fehsel K,Suschek C,et al.Inducible nitric oxide synthasederived nitric oxide in gene regulation,cell death and cell survival〔J〕.Int Immunopharmacol,2001;1:1407-20.
6 Maria Tsoumakidou,Nikolaos Tzanakis,Georgios Chrysofakis,et al.Nitrosative stress,heme oxygenase-1 expression and airway inflammation during severe exacerbations of COPD〔J〕.Chest,2005;127:1911-8.
7 Maestrelli P,Paska C,Saetta M,et al.Decreased haem oxygenase-1 and increased inducible nitric oxide synthase in the lung of severe COPD patients〔J〕.Eur Respir J,2003;21:971-6.
8 Maestrelli P,El Messlemani AH,De Fina O,et al.Increased expression of heme oxygenase(HO)-1 in alveolar spaces and HO-2in alveolar walls of smokers〔J〕.Am J Respir Crit Care Med,2001;164:1508-13.
9 中华医学会呼吸病学分会慢性阻塞性肺疾病学组.慢性阻塞性肺疾病诊治指南〔J〕.中华结核和呼吸杂志,2002;25:453-60.
10 Otterbein LE,Choi AM.Heme oxygenase:colors of defense against cellular stress〔J〕.Am JPhysiol,2000;279:L1029-37.
11 Otterbein LE,Mantell LL,Augustine MK.Carbon monoxide provides protection against hyperoxic lung injury〔J〕.Am J Physiol,1999;276:L688-94.
12 Shinohara T,Kaneko T,Nagashima Y,et al.Adenovirus-mediated transfer and overexpression of heme oxygenase 1 cDNA in lungs attenuates elastase-induced pulmonary emphysema in mice〔J〕.Hum Gene Ther,2005;16(3):318-27.
13 Lee IT,Luo SF,Lee CW,et al.Overexpression of HO-1 protects against TNF-{alpha}-mediated airway inflammation by down-regulation of TNF R1-dependent oxidative stress〔J〕.Am J Pathol,2009;175(2):519-32.
14 Almolki A,Guenegou A,Golda S,et al.Heme oxygenase-1 prevents airway mucus hypersecretion induced by cigarette smoke in rodents and humans〔J〕.Am JPathol,2008;173(4):981-92.
15 Hogg JC,Chu F,Utokaparch S,et al.The natureof small-airway obstruction in chronic obstructive pulmonary disease〔J〕.N Engl JMed,2004;350:2645-53.
16 Rahman I,Biswas SK,Kode A.Oxidant and antioxidant balance in the airways and airway diseases〔J〕.Eur J Pharmacol,2006;533:222-339.
17 Murata M,Kawanishi S.Oxidative DNA damage induced by nitrotyrosine,a biomarker of inflammation〔J〕.Biochem Biophys Res Commun,2004;316:123-8.
18 Ricciardolo FL,Caramori G,Ito K,et al.Nitrosative stress in the bronchina mucosa of severe chronic obstructivepulmonary disease〔J〕.Allergy Clin Immunol,2005;116:1028-35.
19 Rutgers SR,Mark TW,Coers W,et al.Markers of nitric oxide metabolism in sputum and exhaled air are not increased in chronic obstructive pulmonary disease〔J〕.Thorax,1999;54:576-80.
20 Maestrelli P,Paska C,Saettta M,et al.Decreased heme oxygenase-1 and increased inducible nitric oxidesynthasein the lung of severe COPDpatients〔J〕.Eur Respir J,2003;21:971-6.
猜你喜欢
云南医药(2020年5期)2020-10-27
世界科学技术-中医药现代化(2020年2期)2020-07-25
浙江医学(2020年9期)2020-07-01
浙江中西医结合杂志(2019年4期)2019-05-05
浙江医学(2019年2期)2019-01-23
中成药(2018年6期)2018-07-11
天然产物研究与开发(2016年6期)2016-06-05
中国继续医学教育(2015年1期)2016-01-06
医学研究杂志(2015年12期)2015-06-10
癌变·畸变·突变(2015年3期)2015-02-27
