
高效液相色谱法测定食品中的安赛蜜
2011-03-28 06:00:26白静
食品科学 2011年16期
白 静
(北京市通州区产品质量监督检验所,北京 101100)
高效液相色谱法测定食品中的安赛蜜
白 静
(北京市通州区产品质量监督检验所,北京 101100)
建立一种高效液相色谱法(high performance liquid chromatography,HPLC)检测食品中人工合成甜味剂安赛蜜的方法。采用C18反相色谱柱,二极管阵列检测器(diode array detector,DAD),以甲醇∶0.02mol/L乙酸铵溶液=5.5∶94.5(V/V)为流动相,检测波长230nm,流速0.8mL/min,柱温30℃,进样量5μL。安赛蜜出峰时间约在4.6min,样品的检出限为1.7mg/kg,线性范围为1.0~50.0μg/mL,安赛蜜的加标回收率在90%以上,重复性实验的相对标准偏差在2%以下(n=6)。所建立的方法前处理简单,以水为提取溶剂超声波提取安赛蜜,然后在提取液中加入沉淀剂除杂,具有简单、快速、准确、实用性强的特点。
安赛蜜;高效液相色谱;测定
安赛蜜,学名乙酰磺胺酸钾,又名AK糖,是一种非营养型合成甜味剂,分子式为C4H4KNO4S,结构式如图1所示。安赛蜜甜度约为蔗糖的200倍,甜觉快,味质好,无不快后味,稳定性高,耐光、耐热,不致龋齿,适合于糖尿病患者食用,不增加血糖含量[1]。我国GB2760—2007《食品添加剂使用卫生标准》中规定,安赛蜜可用于饮料、冷冻饮品、面包、糕点、糖果、果酱、酱腌菜、蜜饯、胶姆糖、酱油、八宝粥罐头等食品中,并规定了相应的最大使用量[2]。但市场上滥用甜味剂的现象仍时有发生,不法厂商为降低成本赚取利润不择手段,严重违反国家食品添加剂使用卫生标准的相关规定。因此,为防止超标和超范围使用安赛蜜的食品流入市场,危害人身健康,对食品中的安赛蜜进行检测非常有必要。

图1 安赛蜜结构式Fig.1 Chemical structure of acesulfame K
食品中安赛蜜的检测方法主要有高效液相色谱法[3-5]、液-质联用法[6]、离子色谱法[7-8]、薄层色谱法[9]、毛细管电泳法[10-12]等。其中高效液相色谱法是目前最常用的测定安赛蜜的方法,由于食品成分的复杂,选择合适的前处理方法是保证后期液相分析的回收率和重现性的关键;液-质联用法可以大大地降低检测限,提高灵敏度,对组分进行定性和定量的分析,适合于基质复杂的食品分析;离子色谱法因安赛蜜在溶液中以阴离子状态存在,可以使用阴离子交换柱对其进行分离,操作比较简单;薄层色谱法可以用来定性和定量分析安赛蜜,是一种比较快速,价格低廉的技术,但由于薄层板均匀程度和点样的重复性的影响,检测限较低,精确度也不高,应用受到一定的限制;毛细管电泳法样品预处理简单,样品用量少,但因为毛细管柱内径小,进样量少,灵敏度较低[13-15]。
本研究拟建立一种高效液相色谱法检测食品中安赛蜜的方法,采用直接以水为提取溶剂超声波提取安赛蜜,然后加入沉淀剂去除杂质的样品前处理方法,避免复杂的过柱操作,为食品中安赛蜜的检测提供简单、快速、准确、实用的方法。
1 材料与方法
1.1 材料与试剂
安赛蜜标准品 国家标准物质研究中心;甲醇(色谱纯) 美国迪马公司;乙酸铵 北京化工厂;乙酸锌、亚铁氰化钾 北京益利精细化学品有限公司;微孔滤膜(0.45μm) 北京海格里斯公司; 除特别说明,其余试剂均为分析纯;水为蒸馏水。
1.2 仪器与设备
Agilent 1100液相色谱仪(配有DAD检测器) 美国安捷伦公司;AB204-S电子天平 美国Mittler Toledo公司;KQ-500DB超声波清洗器 昆山市超声仪器有限公司。
1.3 方法
1.3.1 样品前处理
1.3.1.1 液体样品
称取5.00g样品于50mL比色管中,加水至约30mL,含二氧化碳样品置于超声波清洗器中超声10min脱气,不含蛋白质样品直接加水定容至刻度,含蛋白质样品依次加入5mL 220g/L乙酸锌溶液、5mL 106g/L亚铁氰化钾溶液,用来沉淀蛋白质等杂质,然后加水定容至刻度,混匀后,用干燥滤纸过滤,滤液经0.45μm微孔滤膜过滤,弃去几滴初滤液后,收集滤液供液相色谱分析。
1.3.1.2 固体样品
称取粉碎均匀的样品5.00g于50mL比色管中,加水至约30mL,摇匀,置于超声波清洗器中超声提取30min以上,以保证样品中的安赛蜜全部溶出,依次加入5mL 220g/L乙酸锌溶液、5mL 106g/L亚铁氰化钾溶液,用来沉淀蛋白质等杂质,然后加水定容至刻度,混匀后,用干燥滤纸过滤,滤液经0.45μm微孔滤膜过滤,弃去几滴初滤液后,收集滤液供液相色谱分析。
1.3.2 色谱条件
色谱柱:Eclipse XDB-C18(4.6mm×150mm,5μm);流动相:甲醇∶0.02mol/L乙酸铵=5.5∶94.5(V/V);检测波长:230nm;柱温:30℃;流速:0.8mL/min;进样量:5μL。
1.3.3 标准曲线
以水为溶剂配制质量浓度为1.0、5.0、10.0、20.0、30.0、40.0、50.0μg/mL安赛蜜标准使用溶液,在1.2.2节色谱条件进行分析,然后以峰面积为纵坐标,以安赛蜜质量浓度为横坐标,绘制标准工作曲线。
1.3.4 试样测定
取经1.2.1节处理后的试样溶液5μL,在1.2.2节色谱条件进行色谱分析,测定其峰面积,从标准曲线查得测定液中安赛蜜的含量。
2 结果与分析
2.1 前处理方法的选择
2.1.1 提取溶剂和提取方法的选择
安赛蜜易溶于水,在20℃溶解度为270g/L,而难溶于乙醇等有机溶剂,20℃时在无水乙醇中的溶解度仅为1g/L,所以选择纯水作为安赛蜜的提取溶剂[16]。常用的样品提取方法有振荡提取法和超声提取法,由于超声提取法能使分子运动加快,促进固液之间的接触,使组分脱附和溶解加快,使提取更快、更彻底,所以选择超声提取法作为安赛蜜的提取方法。以水为提取溶剂,超声波提取30min以上,安赛蜜的加标回收率在91.0%~98.1%之间。
2.1.2 除去干扰物质方法的选择
食品成分复杂,蛋白质等成分会影响安赛蜜的检测,须在前处理中除去这些杂质成分,选用沉淀剂可以较完全地除去干扰物质,选用的沉淀剂要满足以下条件:不吸附或沉淀被测物质安赛蜜,而且应不干扰后面的分析操作,易于除掉。本研究选用乙酸锌和亚铁氰化钾作为蛋白质沉淀剂,这两种试剂混合后形成白色的亚铁氰化锌沉淀,能使溶液中的蛋白质等杂质共同沉淀下来,而不干扰安赛蜜的检测,且容易通过过滤除去。选用乙酸锌和亚铁氰化钾作为蛋白质沉淀剂,安赛蜜的加标回收率可达90%以上。
2.2 检测波长的选择
将安赛蜜的标准溶液进行紫外扫描,得到紫外光谱图,如图2所示。从图2可知,安赛蜜在230nm处有最大吸收,因此确定安赛蜜的检测波长为230nm。

图2 安赛蜜紫外光谱图Fig.2 UV absorption spectrum of acesulfame K
2.3 流动相的选择
安赛蜜在水中的溶解度大,极性强,且呈中性,1%水溶液的pH值为5.5~7.5,本实验选择甲醇-乙酸铵体系作为流动相,获得了峰形尖锐且对称的峰。安赛蜜的出峰时间约在4.6min,安赛蜜标准品和含安赛蜜样品的色谱图分别如图3所示。
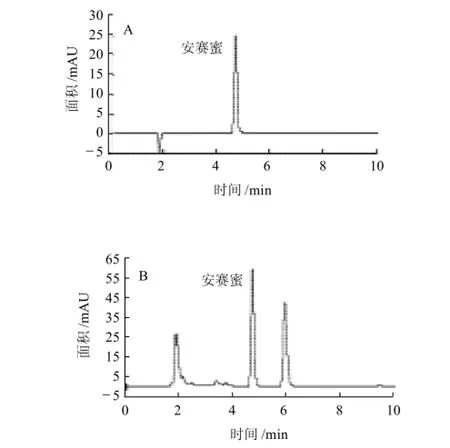
图3 安赛蜜标准品(A)和含安赛蜜样品(B)溶液色谱图Fig.3 HPLC chromatograms of acesulfame K standard (A) and sample (B)
2.4 标准曲线及回归方程
将不同质量浓度的安赛蜜标准溶液分别进样5μL,结果见表1。

表1 安赛蜜标准溶液质量浓度和峰面积Table 1 Acesulfame K concentration versus HPLC peak area
以峰面积为纵坐标,质量浓度为横坐标进行线性回归,得到回归方程为:y=18.336χ+2.2306,相关系数R=1,绘制标准曲线如图4所示。结果表明,安赛蜜在1.0~50.0μg/mL范围内呈良好的线性关系。

图4 安赛蜜标准曲线Fig.4 Standard curve of acesulfame K
2.5 检出限
将低质量浓度的安赛蜜标准溶液进行色谱分析,计算信噪比,在RSN=3时计算检出限,得到安赛蜜的检出限0.17μg/mL,换算成样品的检出限1.7mg/kg。
2.6 回收率和精密度
称取12份5.00g不含安赛蜜的果汁样品,分别加入安赛蜜标准品,作高、低两个质量浓度水平的加标回收实验,每个质量浓度作6个平行测定,结果见表2。

表2 安赛蜜回收率和精密度实验结果Table 2 Mean spike recoveries and relative standard deviation for acesulfame K in a blank juice sample
从表2可以看出,进行6次平行实验,样品的平均加标回收率在90%以上,相对标准偏差在2%以下,本方法的回收率及重现性均很好。
3 结 论
采用高效液相色谱法测定食品中的安赛蜜,使用C18反相色谱柱,以甲醇∶0.02mol/L乙酸铵溶液=5.5∶94.5 (V/V)为流动相,检测波长230nm,流速0.8mL/min,柱温30℃,进样量5μL。确定不同形态食品的前处理方法,以水为安赛蜜的提取溶剂、采用超声波提取方法,提取液通过加入沉淀剂去除杂质,操作简单。并进行方法的确证实验,样品的检出限为1.7mg/kg,线性范围为1.0~50.0μg/mL,安赛蜜的加标回收率在90%以上,重复性实验的相对标准偏差在2%以下(n=6)。结果表明:方法稳定性好,检出限低,重复性好,回收率高。建立的方法用于食品中安赛蜜的分析具有操作简单,分析时间短,准确性好的特点。另外可同时测定食品中的苯甲酸、山梨酸、糖精钠等食品添加剂,是一种实用性很强的检测方法。
[1]阮春梅. 食品添加剂应用技术[M]. 北京∶ 中国农业出版社, 2008∶ 124.
[2]中华人民共和国卫生部, 中国国家标准化管理委员会. GB2760—2007 食品添加剂使用卫生标准[S]. 北京∶ 中国标准出版社, 2008.
[3]黄薇, 刘祥萍. 高效液相色谱法测定食品中安赛蜜[J]. 中国卫生检验杂志, 2007, 17(9)∶ 1624-1625.
[4]肖有玉. 高效液相色谱法测定固体食品中安赛蜜[J]. 江苏食品与发酵, 2007 (2)∶ 39-40.
[5]谢勇. 聚酰胺固相萃取法在食品安赛蜜和糖精钠检测中的应用[J]. 福建轻纺, 2009 (5)∶ 47-49.
[6]李传慧, 李淑娟, 安娟, 等. 快速分离液相色谱/串联四级杆电喷雾质谱法同时测定食品中多种人工合成甜味剂[J]. 食品工业科技, 2009, 30(8)∶ 319-320; 358.
[7]ZHU Yan, GUO Yingying, YE Mingli, et al. Separation and simultaneous determination of four artificial sweeteners in food and beverages by ion chromatography[J]. Journal of Chromatography A, 2005, 1085(1)∶ 143-146.
[8]郭莹莹, 朱岩, 叶明立. 淋洗液发生器离子色谱抑制电导法测定甜味剂[J]. 浙江大学学报∶ 理学版, 2004, 31(4)∶ 435-437.
[9]MAHINDRU S N. Food additives∶ characteristics, detection and estimation[M]. New DelHI∶ APH Publishing Corporation, 2008∶ 88.
[10]THOMPSON C O, TRENERRY V C, KEMMERY B. Micellar electrokinetic capillary chromatographic determination of artificial sweeteners in low-Joule soft drinks and other foods[J]. Journal of Chromatography A, 1995, 694(2)∶ 507-514.
[11]FRAZIER R A, INNS E L, DOSSI N, et al. Development of a capillary electrophoresis method for the simultaneous analysis of artificial sweeteners, preservatives and colors in soft drinks[J]. Journal of Chromatography A, 2000, 876(1/2)∶ 213-220.
[12]蒋奕修, 魏瑞霞, 杨桂珍, 等. 磺胺类人工合成甜味剂的毛细管电泳/电导法分离检测[J]. 分析测试学报, 2009, 28(17)∶ 838- 841.
[13]肖立群, 张承聪, 张承明. 人工合成甜味剂的检测技术研究进展及应用[J]. 云南化工, 2010, 37(4)∶ 49-53.
[14]杨大进, 赵凯, 方从容, 等. 防腐剂和甜味剂检测技术进展[J]. 中国卫生检验杂志, 2008, 18(7)∶ 1460-1463.
[15]鲁琳, 杭义萍, 高燕红, 等. 食品甜味剂分类及其检测技术现状[J].现代预防医学, 2009, 36(11)∶ 2033-2035.
[16]MAYER D G, KEMPER F H. Acesulfame-K[M]. New York∶ Marcel Dekker Inc, 1991∶ 211.
Determination of Acesulfame K in Food by High Performance Liquid Chromatography
BAI Jing
(Beijing Tongzhou Institute of Supervision & Testing on Product Quality, Beijing 101100, China)
A high performance liquid chromatography method for the determination of the artificial sweetener acesulfame K in food was developed. Ultrasonic-assisted aqueous extraction followed by the removal of protein impurities by adding zinc acetate and potassium ferrocyanide was used for sample preparation. Acesulfame K was separated on a C18column using methanol-0.02 mol/L ammonium acetate (5.5∶94.5, V/V) as mobile phase at a flow rate of 0.8 mL/min and detected using a diode array detector (DAD) at 230 nm. The column temperature was set at 30 ℃ and the injection volume 5μL. The retention time of acesulfame K was about 4.6 min. The detection limit of the developed method was 1.7 mg/kg, and the linear range was between 1.0 and 50.0μg/mL. The mean spike recoveries for acesulfame K in a blank juice sample exceeded 90%, with a relative standard deviation below 2% (n=6). In this method, acesulfame K was extracted using water as solvent with the assistance of ultrasound, and purified by precipitation. The method proved to be simple, rapid, accurate and practical.
acesulfame K;high performance liquid chromatography (HPLC);determination
TS207.3
A
1002-6630(2011)16-0245-04
2010-11-03
白静(1978—),女,工程师,硕士,研究方向为食品色谱检测技术。E-mail:chengmin729@yahoo.com.cn
猜你喜欢
应用化工(2022年1期)2022-03-24 13:31:26
环境保护与循环经济(2021年7期)2021-11-02 08:10:52
食品安全导刊(2021年20期)2021-08-30 06:39:50
食品安全导刊(2021年20期)2021-08-30 06:39:22
家庭百事通·健康一点通(2016年9期)2016-09-21 18:42:50
少年科学(2015年10期)2015-10-31 04:19:47
江苏调味副食品(2015年1期)2015-02-28 01:56:34
河北工业科技(2015年4期)2015-02-27 13:15:36
食品工业科技(2014年9期)2014-03-11 18:15:45
中国质量与标准导报(2014年6期)2014-02-28 22:24:10
