
高速逆流色谱法分离前胡提取物中香豆素类成分
2011-03-07 15:00:12蔡海林杜良伟刘祥英陈桂华王彦辉柏连阳
湖南农业大学学报(自然科学版) 2011年1期
蔡海林,杜良伟,刘祥英,陈桂华,王彦辉,柏连阳,3*
(1.湖南农业大学 生物安全科学技术学院,湖南 长沙 410128;2.广西大学 化学化工学院,广西 南宁 530004;3.湖南人文科学技术学院,湖南 娄底 417000)
对白花前胡(Peucedanum praeruptorumDunn.)功能成分的提取主要采用水煎煮、乙醇加热回流法等,利用效率不高[1],而对前胡提取物的分离则主要采用硅胶柱层析、制备液相色谱分离等技术[2],成本较高,制备量小,过程复杂,不适合大规模的分离制备。为了弥补上述方法的不足,笔者采用CO2超临界流体萃取对样品所含香豆素类功能成分进行初提后,用硅胶柱对初提物进行第一步分离,再用高速逆流色谱(HSCCC)[3]对目标成分实现细分,并得到单体化合物。
1 材料与方法
1.1 材 料
前胡购自湖南省药材公司,经湖南省中医药大学于永秩教授鉴定确认为伞形科白花前胡。
主要仪器:HPTLC薄层色谱仪(德国CAMAG公司);HL-1L/40-A型超临界萃取仪(杭州华黎泵业有限责任公司);EYELA旋转蒸发仪(上海爱朗仪器有限公司);岛津 Uvmini-1240紫外分光光度计(日本岛津公司); TBE-300A高速逆流色谱仪(上海同田生化技术有限公司);岛津LC-20A高效液相色谱仪(日本岛津公司); Varian INOVA-300核磁共振仪(瑞士Broker公司)。
1.2 方 法
1.2.1 样品制备
前胡洗净后自然阴干,机械粉碎。取粉碎的前胡根茎2.0 kg,以1∶1的比例用无水乙醇浸泡24 h,采用CO2超临界流体萃取其中的香豆素类成分,萃取温度60 ℃,萃取压力35 MPa,萃取时间60 min,CO2动态流量2 kg/h。
精确称取前胡CO2超临界萃取物10 g(相当于前胡粉碎物200 g),用丙酮溶解后,加入100 g硅胶拌匀;硅胶柱柱径8 cm,柱长120 cm,硅胶1 500 g,石油醚湿法装柱,装柱后反复用石油醚冲洗,湿法上样。利用薄层层析色谱(TLC)试验确定石油醚∶乙酸乙酯体系为硅胶柱分离的洗脱系统。
收集分离组分:每100 mL收集1份,流速控制在0.08 BV/h(100 mL/h),分步收集,以薄层层析结合5%三氯化铁甲醇溶液显色,监测柱层析各分离组分中成分的变化,调整洗脱比例。
1.2.2 高速逆流色谱分离
1) 溶剂系统的选择。根据色谱理论,利用HSCCC分离样品的必要条件是样品在两相中具有合适的分配系数(0.5<K<2.0)[6],可用HPLC、EC、TLC测定[7]。本试验选用HPLC测定,共考察了7个溶剂系统,分别是:正己烷∶乙酸乙酯∶甲醇∶水=1∶1∶1∶1 ,正己烷∶乙酸乙酯∶乙腈∶甲醇=5∶2∶5∶4,正己烷∶乙醇∶水=6∶5,正己烷∶乙腈∶氯仿=5∶5∶1,正己烷∶乙腈=1∶1,石油醚∶乙酸乙酯∶甲醇∶水=5∶5∶5∶4,石油醚∶乙酸乙酯∶甲醇∶水=5∶5∶5∶2。
2) 高速逆流色谱分离初提物。将待测溶剂加入分液漏斗中,充分振摇后静置24 h分相,上相为固定相,下相为流动相,上、下相超声脱气20 min,测定其固定相保留率。考虑到样品浓度高固定相易流失的因素,将样品用等体积的上相和下相溶解,尽可能用大体积稀释样品进样。HSCCC主机转速为850 r/min,流动相流速为2.0 mL/min,254 nm波长检测,根据色谱图手动收集各色谱峰组分。
3) 检测各组分纯度。采用HPLC检测经HSCCC分离后各组分的纯度。HPLC分析条件:流动相为甲醇∶水,梯度洗脱:0 min、0 甲醇→70 min、100%甲醇;流速0.8 mL/min;检测波长为254 nm,柱温30 ℃ 。
1.2.3 鉴定单体化合物结构
将分离所得单体化合物冷冻干燥,利用核磁共振(NMR)检测并鉴定其结构。
2 结果与分析
2.1 初提物的初步分离
超临界 CO2萃取白花前胡甲素提取率平均为2.66%,高于采用乙醇加热回流法1.81%的提取率。根据香豆素类化合物已知的各种理化特征,以石油醚∶乙酸乙酯体系为洗脱系统,硅胶柱分离共收集520个组分,经过TLC检测,合并相同组分,用旋转蒸发仪浓缩,共得到14种分离组分,其中A2、A4、A6、A13组分在365 nm紫外灯照射下呈亮蓝色,具备香豆素类成分的显著特征[8],将这4个组分混合后作为样品(记为B1)进行HSCCC分离。
2.2 高速逆流色谱分离结果
2.2.1 溶剂系统的确定
根据表1的K值结果,选择石油醚∶乙酸乙酯∶甲醇∶水= 5∶5∶5∶4作为溶剂体系进行 HSCCC分离。该溶剂体系HPLC色谱图见图1、图2。
2.2.2 HSCCC分离功能成分
选择石油醚∶乙酸乙酯∶甲醇∶水=5∶5∶5∶4作为溶剂体系进行HSCCC分离,共得到4个组分(图3),通过HPLC检验,化合物 Ⅰ (12.0 mg)、化合物 Ⅱ (10.5 mg)、化合物III(5.8 mg)、化合物IV(4.6 mg)纯度分别达到了97.6%、98.1%、95.8%、95.2%,可以用作单体化合物的结构鉴定。
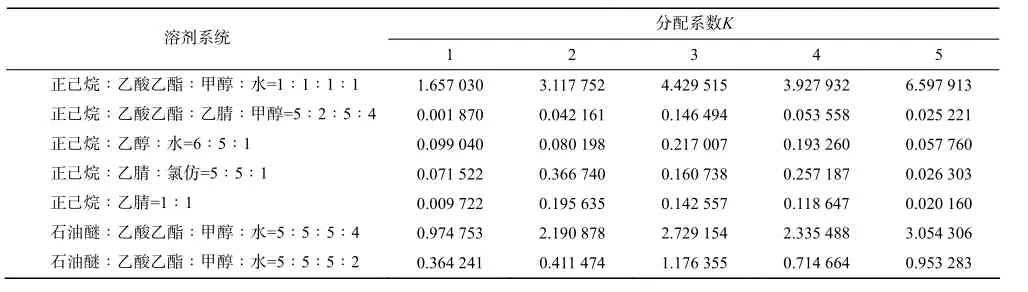
表1 各溶剂体系K值Table 1 K values of different solvent system
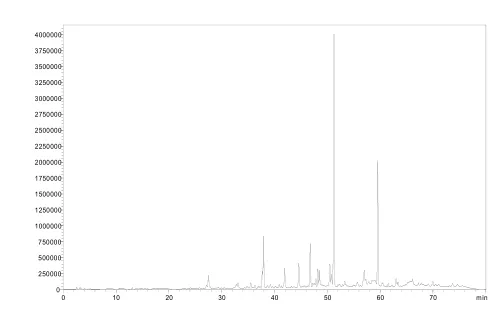
图1 上相溶解样品液相色谱Fig.1 Chromatogram of sample distribution in upper solvent used for HSCCC by HPLC
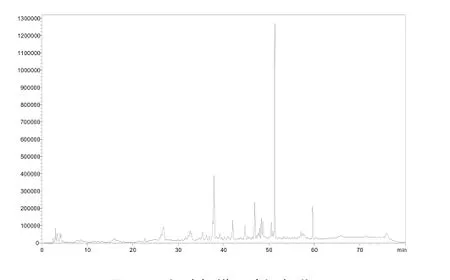
图2 下相溶解样品液相色谱Fig.2 Chromatogram of sample distribution in lower solvent used for HSCCC by HPLC

图3 HSCCC分离效果Fig.3 HSCCC chromatogram
2.3 结构鉴定
通过核磁共振(NMR)对4种化合物进行结构鉴定,解析结果如下。
化合物Ⅰ:1HNMR(300MHz,CDCl3) δppm: 7.61 (1H,d,J=9.6Hz),7.38(1H,d,J=8.7Hz),6.83(1H,d,J=8.7Hz),6.28 (1H,d,J =5.1Hz), 6.23(1H,d,J=9.6Hz),6.07(1H,br.q,J=7.2Hz),5.33(1H,d,J=5.1Hz),2.11(3H,s),2.01(3H,d,q,J= 7.2,1.8Hz),1.84 (3H,t,J=1.5Hz),1.46(3H,s) ,1.39(3H,s);13CNMR(300 MHz,CDCl3)δ ppm: 169.4,166.2,159.8,156.5,154.2,143.2,138.9,129.1,127.1,114.5,113.2,112.5,106.7,77.1,71.7,63.0,23.8,23.5,20.7,20.4,15.7。
该化合物与已知化合物前胡香豆素Ⅱ的波谱数据[9]一致,确定化合物Ⅰ为前胡香豆素Ⅱ。
化合物Ⅱ:1HNMR(300MHz,CDCl3)δ ppm: 7.61(1H,d,J=9.6Hz),7.38(1H,d,J=8.4Hz),6.82(1H,d,J= 8.4Hz),6.63 (1H,d,J=5.1Hz),6.21 (1H,d,J= 9.6Hz),6.02(1H,br.,q,J=7.2Hz),5.34(1H,d,J= 5.1Hz),2.09(3H,s),2.00(3H,br.d,q,J=7.2,1.8Hz),1.87 (3H,m,J=1.5 Hz),1.47(3H,s),1.44 (3H,s);13CNMR (300MHz,CDCl3)δ ppm: 169.8,166.9,159.7,156.6,154.0,143.2,137.7,129.2,127.4,114.4,113.2,112.5,107.2,77.2,70.5,60.1,25.3,22.1,20.7,20.3,15.5。综合分析化合物Ⅱ为北美芹素[10-11]。
化合物III:1H-NMR(300MHz,CDCl3)δ ppm:6.24(1H,d,J=9.6Hz,3-H),7.60(1H,d,J=9.6Hz,4-H),7.35(1H,d,J=8.7Hz,5-H),6.80 (1H,d,J= 8.7Hz,6-H),1.479 (3H,s,2’-CH3),1.437 (3H,s,2’-CH3),5.41(1H,d,J= 4.8Hz,3’- H),2.11 (3H,s,2’’-CH3),6.13(1H,m,3’’’-H),1.87 (3H,m,4’’’-CH3),1.96(3H,d,q,5’’’- CH3);13C-NMR(300 MHz,CDCl3)δ ppm:159.8 (C-2),113.1(C-3),143.2(C-4),129.1 (C-5),114.3 (C-6),156.7 (C-7),107.0(C-8),153.9 (C-9),112.4 (C-10),77.6(C-2’),69.7 (C-3’),61.0(C-4’),24.8(C-2’-CH3),22.9 (C-2’-CH3),169.8(C-1’’),20.6(C-2’’),166.4 (C-1’’’),126.9(C-2’’’),139.8(C-3’’’),15.7 (C-4’’’),20.4(C-5’’’)。综合分析化合物III为白花前胡甲素[12]。
化合物IV:1H-NMR(300MHz,CDCl3)δ ppm:6.37 (1H,d,J=9.6Hz,3-H),7.79(1H,d,J=9.3Hz,4-H),7.69(1H,s,5-H),7.48(1H,s,8-H),7.70(1H,d,J=2.1Hz,2’-H),6.83(1H,dd,J=2.4Hz,3’-H);13C-NMR (300MHz,CDCl3) δ ppm:161.0(C-2),114.6(C-3),144.0(C-4),119.8(C-5),124.8 (C-6), 156.4(C-7),99.8 (C-8),152.0 (C-9),115.4 (C-10),146.8(C-2’),106.3(C-3’)。综合分析化合物IV为补骨脂素[13]。
[1] 刘爱东,侯微,陈雪松,等.CO2超临界流体萃取前胡药材中白花前胡甲素[J].中西医结合学报,2008,12(6):1286-1289.
[2] 陈桂华.前胡提取物诱导水稻抗稻瘟病和抗寒性研究[D].长沙:湖南农业大学生物安全科学技术学院,2007:25-39.
[3] Ito Y,Sandlin J,Bowers W G.Hig-speed preparative counter-current chromatography with a coil Planet centrifuge [J].J Chromatogr,1982,244:247-258.
[4] Wang X,Wang Y Q,Geng Y L,et al.Isolation and purification of honokiol and magnolol from cortexMagnoliae officinalisby high-speed counter-current chromatography [J].J Chromatogr A,2004(2):171-175.
[5] Jiang Y,Lu H T,Chen F.Preparative purification of glycyrrhizin extracted from the root of liquorice using high-speed counter-current chromatography[J].J Chromatogr A,2004(1):183-196.
[6] 丛浦珠,李笋玉.天然有机质谱学[M].北京:中国医药科技出版社,2003:565-566.
[7] Ito Y.Golden rules and pitfalls in selecting optimum conditions for high speed counter-current chromatography [J].J Chromatogr A,2005(2):145-168.
[8] 卢艳花.中药有效成分提取分离实例[M].北京:化学工业出版社,2007:188.
[9] Takata M S,Okuyama,T Structures of angular pyranocoumarins of Bai-Hua Qian-Hu,the root ofPeucedanump raeruptorum[J].Planta Medica,1990,56(3):307-311.
[10] 黄平.南岭前胡素A的结构鉴定[J].药学学报,1997,32(1):62-64.
[11] 孔令义,裴月湖,李铣,等.凯林内酯类香豆素的研究进展[J] .天然产物研究与开发,1994,6(1):50-65.
[12] 陈政雄,黄宝山,佘其龙,等.中药白花前胡化学成分的研究——四种新香豆素的结构[J].药学学报,1979,14 (8):485-489.
[13] 彭国平,吴盘华,李红阳,等.补骨脂化学成分的研究[J].中药材,1996,19 (11):563-565.
英文编辑:易来宾
猜你喜欢
佳木斯大学学报(自然科学版)(2020年1期)2020-02-28 05:30:52
农药科学与管理(2019年8期)2019-11-23 08:04:44
天然产物研究与开发(2018年10期)2018-11-06 07:43:42
天然产物研究与开发(2018年6期)2018-07-09 06:01:46
天然产物研究与开发(2018年2期)2018-04-04 02:01:34
中成药(2017年12期)2018-01-19 02:06:26
中成药(2017年4期)2017-05-17 06:09:46
农产品加工(2017年6期)2017-05-09 18:04:52
核科学与工程(2015年3期)2015-09-26 11:58:24
天然产物研究与开发(2014年6期)2014-04-27 14:15:54
