
原位制备聚己内酯和羟基磷灰石复合材料
2011-03-05 00:43徐子颉逯爱慧
同济大学学报(自然科学版) 2011年10期
徐子颉,马 超,汪 飞,逯爱慧
(同济大学 化学系,上海200092)
使用聚己内酯(PCL)和羟基磷灰石(HAP)制备出的PCL/HAP复合材料,具有优良的机械特性、生物相容性以及在人体内可生物降解的特性,可用于诱导骨细胞的生长,人体骨骼修复的支架材料以及用于临床治疗骨感染等疾病的药物载体和药物控释材料[1-5].聚己內酯是一种无毒无害,在人体内可降解,生物相容性好的医用生物材料;但是,由于聚己內酯具有结晶性较强,在体内降解速率过慢的特性,使得该材料在生物医学领域的应用范围受到限制.羟基磷灰石具有良好的生物活性,但是它具有较大的脆性而且在体内的生物降解速率较快[6-11].通过将这2种物质进行复合,可以充分发挥二者各自的材料特性,得到符合临床生物医学材料要求的新型复合材料.
目前,PCL/HAP复合材料的制备方法主要包括共混法和 HAP表面共聚法[12-19].共混法是将预先制备的HAP粉体与PCL熔液共混,此方法制备工艺简单,成本较低,但是通过该方法制得的PCL/HAP复合材料中,PCL和HAP组分间存在明显的相分离现象,两组分各自的材质特性依然突显;共聚法通过在HAP表面羟基基团处引发己内酯(ε-CL)开环聚合,制备了PCL/HAP复合生物材料.虽然该方法能较好地解决PCL与HAP之间的相分离问题,但是,该制备工艺复杂,反应时间较长,生产成本高,只适合HAP粉体表面包覆改性,而不适用于块状PCL/HAP复合材料的制备.
本文采用HAP与PCL的溶胶-凝胶技术和真空干燥技术,在四氢呋喃有机溶剂体系中原位合成了PCL/HAP多孔复合材料.研究了在溶胶-凝胶过程中,HAP在其成核过程中与PCL之间的弱氢键作用,探讨了凝胶体系中网络结构的形成,以及对羟基磷灰石晶体颗粒生长的影响.
1 实验部分
1.1 实验材料
四水合硝酸钙(Ca(NO3)2·4H2O,分析纯,国药集团化学试剂),浓磷酸(H3PO4,分析纯,质量分数85%,国药集团化学试剂),浓氨水(NH4OH,分析纯,质量分数25%~28%,国药集团化学试剂),聚已内酯((C6H8O4)n,平均相对分子质量70 000,日本大赛璐化学工业),四氢呋喃(C4H8O,分析纯,国药集团化学试剂),油酸 (C18H34O2,分析纯,山东省济南云翔化工).
1.2 PCL/HAP复合材料的制备
按Ca与P的原子数量比为1∶1.7,控制四水合硝酸钙和浓磷酸的比例,以浓氨水调节体系的pH值;按HAP在PCL/HAP复合材料中的质量分数分别为20%,40%,60%和80%的比例,制备得到一系列样品,分别记为HP20,HP40,HP60和HP80.在室温条件下,取20 mL的四氢呋喃,向其中加入浓磷酸并搅拌15 min,再取10 mL的四氢呋喃,向其中加入3 mL浓氨水并搅拌15 min,然后在搅拌条件下将上述溶液混合并继续搅拌30 min;用氨水调节混合溶液的pH值大于10,然后加入四水合硝酸钙,使其均匀分散,随后向其中加入1.5 mL油酸,继续搅拌并加入PCL,静置后得到凝胶,再经洗涤和真空干燥,获得PCL/HAP复合材料.
1.3 复合材料的表征
使用inVia显微共聚激光拉曼光谱仪,采用连续扫描的形式,选择785激光器,在100~4 000cm-1波数段扫描,测定样品的拉曼光谱.使用SSX-550型扫描电子显微镜观测样品的形貌.使用D8型X射线粉末衍射仪(CuKa,步长0.02°扫描速率12(°)·min-1,扫描范围10°~70°)测定样品的 X射线衍射图谱.
2 结果与讨论
本文中HAP形成的反应式如下:
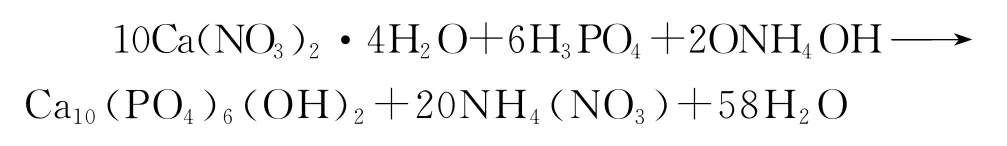
图1是一系列PCL/HAP复合材料的XRD(X射线粉末衍射仪)谱图,从中可以分别观察到PCL和HAP物相的特征衍射峰,表明采用本文的制备方法得到了PCL/HAP复合物.从图中可见,随着HAP在复合材料中含量的增加,其特征峰的相对强度逐渐增强,与此同时,PCL的特征衍射峰的相对强度显著降低,表明其结晶程度随HAP含量的增加而下降.在溶胶-凝胶过程中,PCL和HAP无机分子在纳米尺度均合接触,提高了HAP在PCL中的稳定性和分散性,降低了PCL的结晶程度,从而在一定程度上避免了相分离现象的出现.

图1 PCL/HAP复合物的XRD衍射图谱Fig.1 XRD patterns of PCL/HAPs composite
图2是一系列复合材料以及纯的PCL样品的拉曼光谱.从图中可见,随着复合材料中PCL含量的减少,酯基基团的平面振动、亚甲基面内变形振动、骨架伸缩振动,以及亚甲基的非对称弯曲振动强度逐渐降低.与此同时,磷酸根的v3振动、v1振动、随复合材料中HAP含量的增加,其相对强度逐渐增强,另外,亚甲基链的非对称伸缩振动的相对强度递减,而且拉曼光谱的谱峰出现宽化,这说明复合材料中PCL的结晶程度逐渐降低.在纯PCL样品中,于1 654cm-1处不出峰,但在复合材料样品中,随着HAP含量的增加,该处的谱峰强度逐渐增强,表明PCL与HAP分子间存在弱氢键作用.这是因为,PCL在凝胶体系中形成的网络结构限制了HAP分子的扩散范围,降低了HAP分子在结晶过程中的浓度梯度,延缓了HAP晶粒的生长速率,在体系中形成了纳米级的HAP晶粒,使其具有较高的比表面能,极易与PCL分子形成弱氢键作用.在2 866cm-1处分别为亚甲基链的对称伸缩振动,其波数随复合材料样品中HAP含量的增加向波数增大的方向发生位移.这是因为HAP的加入使PCL的分子运动受到限制,使PCL的长链分子在震动过程中需要更大能量,因此波数向高频方向移动.进一步表明HAP中的羟基基团与PCL分子中的酯基形成氢键,这种相互作用使PCL高聚物分子牢牢固定在HAP分子表面,一方面限制了HAP晶体的进一步长大,另一方面使PCL分子链段在运动中受限,限制了聚己内酯的结晶行为,导致PCL的结晶度显著降低,使得复合材料的均匀性和稳定性显著增强.目前使用共混法制备得到的PCL/HAP复合材料中,HAP晶体多为微米级,晶体自身体积较大,很难与PCL分子充分接触,形成氢键,因此,两物相依然保持着各自的材质特性.
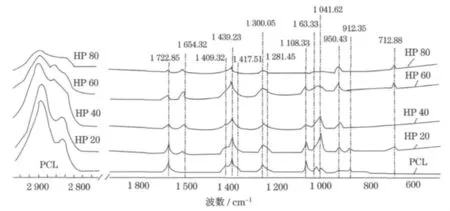
图2 PCL/HAP复合物的拉曼光谱Fig.2 Raman spectrum of PCL/HAPs composite
图3是PCL/HAP复合材料的SEM(扫描电镜)照片,正如图3所示,其中图a和图b分别为HAP组分在PCL中质量分数为20%和40%时样品的形貌图.从图中可看出整个体系混合均匀,箭头所指处没有观察到有相分离现象的出现.图中的孔洞是PCL/HAP凝胶在真空干燥过程中溶剂挥发后所致.图c和图d分别为当HAP组分在PCL中的质量分数为60%和80%时的电镜形貌图,从箭头所指处可看出体系中发生明显的相分离.因为过量的HAP分子使其局部的结晶饱和度过大,从而形成了较大的HAP晶粒,影响了HAP在PCL体系中的稳定性.

图3 PCL/HAP复合材料的SEM照片Fig.3 SEM morphology of PCL/HAP samples
3 结论
在四氢呋喃分散系中通过溶胶-凝胶技术和真空干燥技术原位合成出了HAP质量分数在40%以下仍能均匀分散的PCL/HAP复合材料;通过在体系中加入油酸来防止PCL结晶,提高了复合材料的稳定性;PCL分子对HAP晶核生长有一定的限制,阻碍了HAP晶体颗粒的长大;纳米级的HAP晶体与PCL之间存在明显的氢键作用,使PCL分子链段在运动中受限,大大降低了PCL的结晶程度.
[1] Neumann M,Epple M.Composites of calcium phosphate and polymers as bone substitution materials[J].European Journal of Trauma,2006,32(2):125.
[2] Oki A,Qiu X,Olajide A.Synthesis of organic-inorganic hybrid composite and its thermal conversion to porous bioactive glass monolith[J].Elsevier Science Materials,2006,60(21):2751.
[3] Kikuchi M,Suetsugu Y,Tanaka J,et al.Preparation and mechanical properties of calcium phosphate/copoly-L-lactide composites[J].Journal of Materials Science:Materials in Medicine,1997,8(6):361.
[4] Zhang R,Peter X,Ma B.Polymer/apatite composite scaffolds for mineralized tissue engineering[J].Macromd Biosci,2004,4(2):100.
[5] Marra K G,Jeffrey W,Szem P,et al.In vitro analysis of biodegradable polymer blend/hydroxyapatite composites for bone tissue engineering[J].Biomedical Material Research,1999,47(3):324.
[6] Rhee S H,Choi J Y,Kim H M.Preparation of a bioactive and degradable poly(-caprolactone)/silica hybrid through a sol-gel method[J].Biomaterials,2002,23(16):4915.
[7] Ma P X.Biomimetic materials for tissue engineering[J].Advanced Drug Delivery Reviews,2008,60:184.
[8] Liu X H,Ma P X.Polymeric scaffolds for bone tissue engineering[J].Annals of Biomedical Engineering,2004,32(3):477.
[9] Doremus R H.Bioceramics:a review [J].Journals of the Minerals,Metals and Materials Society,1992,27(3):285.
[10] Guerra G D,Cerrai P,Tricoli M.Composites between hydroxyapatite and poly(ε-caprolactone)synthesized in open system at room temperature[J].Journal of Materials Science:Materials in Medicine,2006,17(1):69.
[11] Roeder R K,Converse G L.Hydroxyapatite-reinforced polymer biocomposites for synthetic bone substitutes[J].Journals of the Minerals,Metals and Materials Society,2008(3):35.
[12] Varma H K,Sivakumar R.Dense hydroxy apatite ceramics through gel casting technique[J].Materials Letters,1996,29:57.
[13] Ramesh S,Tan C Y,Sopyan I,et al.Consolidation of nanocrystalline hydroxyapatite powder [J].Science and Technology of Advanced Materials,2007,8(1):124.
[14] Natarajan U V,Rajeswari J.Influence of calcium precursors on the morphology and crystallinity of sol-gel-derived hydroxyapatite nanoparticles[J].Crystal Growth,2008,310(21):4601.
[15] Rajabi-Zamani A H,Behnamghader A,Kazemzadeh A.Synthesis of nanocrystalline carbonated hydroxyapatite powder via nonalkoxide sol-gel method [J].Materials Science and Engineering,2008,28(8):1326.
[16] Li Y,Tjandra W,Tam K C.Synthesis of amorphous calcium phosphate using various types of cyclodextrins[J].Materials Research Bulletin,2008,42(5):2318.
[17] Tseng Y H,Kuo C S,Li Y,et al.Polymer-assisted synthesis of hydroxyapatite nanoparticle [J].Materials Science and Engineering C,2009,29(3):819.
[18] Balamurugan A,Balossier G,Torres P,et al.Sol-gel synthesis and spectrometric structural evaluation of strontium substituted hydroxyapatite[J].Materials Science and Engineering C,2009,29(3):1006.
[19] Abeer M,Kady E,Khaled R,et al.Characterization and bioactivity evaluation of calcium pyrophosphate/polymeric biocomposites[J].Ceramics International,2009,35(7):1.
猜你喜欢
化工设计(2022年4期)2023-01-02
贵州科学(2022年4期)2022-09-05
高中数理化(2022年14期)2022-08-15
天津中医药(2020年5期)2020-06-01
原子与分子物理学报(2019年5期)2019-04-28
化工管理(2017年35期)2018-01-10
中成药(2017年4期)2017-05-17
中国民族医药杂志(2016年6期)2016-05-09
中学化学(2015年12期)2016-01-19
应用化工(2014年5期)2014-08-08