
灵杆菌非特异性核酸酶的原核表达、纯化及活性分析
2011-02-09 09:31陈鹏杨海艳李慧婧杨龙雨李学俊
生物工程学报 2011年8期
陈鹏,杨海艳,李慧婧,杨龙雨,李学俊
西北农林科技大学生命科学学院,杨凌 712100
重组蛋白质表达技术的成熟与发展为研究蛋白质的结构与功能提供了高效的研究手段,同时重组表达技术为众多蛋白质与酶的大规模制备和应用奠定了基础。但目前在重组蛋白特别是医用重组蛋白纯化过程中还面临诸多问题,如菌体释放的核酸会产生高粘度的裂解液进而影响下游的操作和层析纯化,同时过量的核酸将导致纯化产品的核酸污染等。核酸的污染也会影响到纯化过程蛋白的定量以及活性分析甚至影响蛋白的结晶。现今最常用的核酸去除方法利用了核酸带负电荷的特征,在初步纯化时利用如STREAMLINE SP、SP Sepharose Big Beads、SP或CM Sepharose FF、SP Sepharose XL等离子交换介质除去大量核酸,也有研究利用疏水层析介质去除核酸。但以上方法成本较高,需要昂贵的仪器设备,不适合纯化前期大规模的核酸去除。酶催化的高效性使酶法去除核酸成为理想的途经,已有通过加入核酸酶去除蛋白样品中核酸的研究实例。如商业化产品 Benzonase endonuclease、DNase和RNase A等,其中Benzonase endonuclease来源于灵杆菌。灵杆菌核酸酶 (Serratia marcescens nuclease,smNU) 是一种分泌至胞外的非特异性磷酸二酯酶,其在金属阳离子存在下可以高效切割任意形式的核酸 (DNA和RNA,双链和单链)[1-5],其催化效率是葡萄球菌核酸酶的4倍,牛胰DNase I的34倍。灵杆菌是一种条件致病菌,致使大规模培养菌液用于核酸酶的分离纯化存在潜在的危险,因此有必要探索该酶安全高效的重组表达体系。由于核酸酶的胞内表达可能造成重组表达宿主细胞自身核酸的降解而产生宿主细胞的强毒性,因此已报道的 smNU重组表达均采用胞外分泌的方式,但表达效率较低,每升仅可生产1.23×104U的酶[6]。本研究在成功克隆了 smNU基因的基础上,构建了smNU的胞内原核表达载体,实现了该酶的胞内高效表达,并用Amylose resin纯化该酶得到了理想的纯化效果,为今后smNU的分子改造以及应用奠定了基础。
1 材料与方法
1.1 材料
1.1.1 菌种与质粒
经鉴定的灵杆菌为本实验室保存,大肠杆菌BL21感受态细胞购自北京全式金生物技术有限公司。pMAL-c4X载体为New England Biolabs公司产品,其分子大小为6 645 bp,编码麦芽糖结合蛋白的片段大小为1 388 bp。
1.1.2 工具酶和主要试剂
实验用到的限制性核酸内切酶、T4 DNA连接酶、蛋白分子质量Marker均购自Fermentas公司。Pfu Turbo DNA聚合酶为 Stratagene公司产品。DNA Marker购自大连宝生物工程有限公司。其他常用生化及分子生物学试剂均为国产或进口分析纯。Amylose resin为NEB公司产品。引物合成及测序由北京奥科生物技术有限责任公司完成。
1.2 方法
1.2.1 灵杆菌核酸酶原核表达载体的构建
根据smNU的基因序列设计并合成引物。上游引物 NU12-1: 5′-GCCGACACGCTCGAATCCAT C-3′,下游引物 NU12-2: 5′-AGTCGGATCC TCAG TTTTTGCAGCCCATCAACTCC-3′,下游引物引入BamHⅠ的酶切位点 (下划线所示)。以灵杆菌的基因组DNA为模板,进行PCR扩增。PCR反应体系为 (25 µL):1×PCR缓冲液,2 mmol/L MgCl2,0.2 mmol/L dNTPs,75 ng灵杆菌基因组DNA,200 nmol/L上下游引物,1 U Pfu Turbo DNA聚合酶。PCR反应参数为:95 ℃ 3 min;95 ℃ 20 s,61 ℃30 s,72 ℃ 1 min ,25个循环;最后72 ℃延伸7 min。 PCR产物采用 PCR产物回收试剂盒纯化后进行BamHⅠ酶切,质粒 pMAL-c4X采用 XmnⅠ和BamHⅠ双酶切,胶回收大片段后进行连接,连接产物转入E. coli BL21。经菌落PCR验证为阳性的克隆,提取质粒送北京奥科生物技术有限责任公司进行测序鉴定。
1.2.2 序列分析
将测序所得的序列在 NCBI网站 (http://www. ncbi.nlm.nih.gov/blast),采用 BLASTN软件进行相似性搜索,核酸序列同源性比较用ClustalX2程序进行分析。
1.2.3 MBP-NU融合蛋白的诱导表达
将构建正确的重组质粒pMAL-c4X-NU转入E. coli BL21,37 ℃培养7 h后,用于扩大培养。
1) 诱导温度和IPTG浓度对MBP-NU融合蛋白表达量的影响:将扩大培养的菌液按1∶100接种于LB液体培养基中,分别置于24 ℃、28 ℃、37 ℃培养至菌浓度OD600值为1.0时,分别加入IPTG至终浓度为0.5、0.75、1.0 mmol/L,诱导培养3 h,收获细菌培养物,SDS-PAGE分析表达情况,分离胶浓度为12.5%。
2) 诱导时间对 MBP-NU融合蛋白表达量的影响:将扩大培养的菌液按1∶100接种于LB液体培养基中,37 ℃培养至菌浓度OD600值为1.0时,加入IPTG至终浓度为0.75 mmol/L,诱导时间分别为0.5、1.5、2.5和3.5 h,收获细菌培养物,SDS-PAGE分析表达结果。
1.2.4 融合蛋白的纯化
融合蛋白的纯化参考NEB公司pMAL试剂盒说明进行[7]。将扩大培养的菌液按 1∶100接种于250 mL LB培养基中,37 ℃培养至菌浓度OD600值为1.0时,加入IPTG至终浓度为0.75 mmol/L,诱导培养2 h后,8 000 r/min离心10 min并将收获的菌体重悬于40 mL的裂解液 (10 mmol/L Tris-HCl,pH 7.2) 中,−20 ℃放置过夜后,冰浴条件下超声破碎20 min,超声功率为30 W,超声间隔时间为10 s,裂解的菌液12 000 r/min离心10 min,收集上清备用。用8倍柱体积的平衡缓冲液 (10 mmol/L Tris-HCl,pH 7.2) 平衡Amylose resin亲和层析柱,然后加入样品并使其缓慢的流过层析柱以便样品充分的结合,用 10倍柱体积的洗涤缓冲液 (10 mmol/L Tris-HCl,1 mol/L NaCl,pH 7.2) 洗去未结合的蛋白,最后用洗脱缓冲液 (10 mmol/L Tris-HCl,10 mmol/L麦芽糖,pH 7.2) 洗脱,洗脱蛋白用SDS-PAGE进行分析,蛋白的纯度采用Bandscan软件进行分析。
1.2.5 MBP-NU融合蛋白的活性检测
以 10 kb的标准质粒为底物,不同单位的MBP-NU对其进行水解反应。酶活单位的定义:在37 ℃条件下30 min完全降解5 µg DNA的酶量为一个酶活单位。在冰浴上每支PCR管中加入3 µL缓冲液N (250 mmol/L pH 8.0 Tris-HCl,0.5 g/L BSA,5 mmol/L MgSO4),过量的标准质粒,一定量的MBP-NU,用ddH2O补充至终体积为15 µL,混匀,37 ℃保温30 min。1%琼脂糖凝胶电泳检测反应结果。MBP-NU的量分别为:0、0.25、0.5、0.75、1.0、1.25、1.5、2.0、3.0、4.0、5.0、10.0 U。
以含 RNA的 10 kb标准质粒为底物,进行MBP-NU降解 RNA的定性实验。反应体系:3 µL缓冲液N,过量的标准质粒,适量的MBP-NU,再加入ddH2O至终体积15 µL,混匀,37 ℃保温30 min。1%琼脂糖凝胶电泳检测反应结果。MBP-NU的量分别为:0、8、16、24、32、40、48、56 U。
1.2.6 EDTA和PMSF对MBP-NU活性的影响
在下述反应体系中分别加入不同浓度的抑制剂EDTA或PMSF,混匀,37 ℃保温30 min。1%琼脂糖凝胶电泳检测反应结果。EDTA的浓度分别为:0、0.5、1.0、2.0、3.0、4.0、5.0 mmol/L。PMSF的浓度分别为:0、0.5、1.0、1.5、2.0、3.0、4.0、5.0 mmol/L。反应体系为:3 µL缓冲液N,过量的标准质粒,7.5 U的MBP-NU,适量的抑制剂,再加入ddH2O至终体积15 µL。
1.2.7 温度、pH和盐浓度对MBP-NU活性的影响
参考1.2.5的方法,分别在0 ℃、4 ℃、16 ℃、24 ℃、37 ℃、52 ℃、67 ℃、82 ℃条件下检测MBP-NU的水解活性,分析温度对MBP-NU水解活性的影响。用pH分别为3.7、4.5、5.6、7.0、8.0、9.0、10.0的缓冲液替代缓冲液N,在37 ℃条件下检测MBP-NU的水解活性,分析pH对MBP-NU水解活性的影响。
为探讨盐浓度对MBP-NU水解活性的影响,参考1.2.5的方法,在反应体系中加入终浓度分别为0、50、100、150、200、250、300 mmol/L KCl或NaCl,37 ℃条件下检测酶的水解效率。
1.2.8 纯化MBP-NU中蛋白酶活性的检测
纯化蛋白中蛋白酶活性的测定参考 Cupp-Enyard[8]的方法。灭菌试管加入5 mL 0.65% (W/V)酪蛋白溶液,37 ℃水浴中保温10 min,加入2 000 U的纯化酶液,摇匀后,37 ℃精确反应1 h,然后立即向管内加入5 mL 0.4 mol/L三氯乙酸溶液以终止反应。37 ℃水浴中继续保温30 min,12 000 r/min离心20 min,取上清在275 nm波长处测定光吸收。以含有高温灭活酶液的测定管作为对照,每次测定重复3次。
另外分别以商品化的牛血清白蛋白 (Bovine Serum Albumin,BSA) 和本实验室纯化的无机焦磷酸酶 (pyrophosphatase,PPA) 作为反应底物,与10 000 U高剂量的MBP-NU在37 ℃条件下反应24 h后,SDS-PAGE分析BSA和PPA电泳带型的变化。
1.2.9 纯化MBP-NU与商品化Benzonase的活性比较
为了比较MBP-NU与商品化的Benzonase的水解效率,参考1.2.5的方法,在反应体系中分别加入终浓度为1.0、10 U的酶,37 ℃条件下检测酶的水解效率。
1.2.10 纯化MBP-NU的去核酸效果检测
采用内标法检测MBP-NU对核酸的水解效果。本实验室在无机焦磷酸酶的重组表达与纯化实验中,6 g诱导表达的菌体用15 mL裂解液 (25 mmol/L Tris-HCl,50 mmol/L KCl,0.5% Tween20,pH 8.0)进行裂解,取裂解液加入1.2.1中的PCR产物至终浓度为100 ng/µL,再分别添加终浓度为0、1、5、10、15、20 U/mL的MBP-NU,37 ℃条件下反应5 min,置于冰上。取2 µL做模板参考1.2.1扩增参数及体系检测内标水解程度。
2 结果
2.1 灵杆菌核酸酶原核表达载体的构建

图1 NU基因的PCR扩增 (A) 和重组质粒的PCR鉴定 (B)Fig. 1 PCR amplification of NU gene (A) and identification of recombinant vector pMAL-c4X-NU (B). M: DNA marker DL2000; 1: PCR product of NU gene.
以灵杆菌的基因组DNA为模板,PCR扩增编码灵杆菌核酸酶的 DNA片段,电泳分析显示在750 bp左右有一特异DNA带 (图1A),大小与预期结果一致。将扩增产物酶切纯化后连接到 pMAL-c4X载体上,得到重组表达载体pMAL-c4X-NU。提取重组表达载体,质粒PCR扩增得到约750 bp的基因片段 (图1B),与目的序列大小一致,并经测序证实载体构建完全正确。
2.2 灵杆菌核酸酶的序列分析
用BLASTN在线软件对所测NU序列进行同源基因检索显示其与 S. marcescens 核酸酶基因的同源性为97%,与Serratia proteamaculans 568的核酸酶基因的同源性为 85%。应用 ClustalX2软件对其核酸序列进行比对发现其与S. marcescens核酸酶基因 (GenBank Accession No. M19495.1) 有22个碱基不同,编码的氨基酸仅在第12位不同,其他均为同义密码子的变化 (图2)。

图2 灵杆菌核酸酶NU和与其同源的核酸序列比对Fig. 2 Alignment of nucleic acids between NU and its homologous sequences. The reading frame of the maturation protein was highlighted by gray coating.
2.3 MBP-NU融合蛋白的诱导表达
SDS-PAGE结果显示,经IPTG诱导的含重组表达载体的细菌可大量表达大小约为 78 kDa的新蛋白,而分别含有 pMAL-c4X、pMAL-c4X-NU (未经IPTG诱导) 载体的宿主菌及不含表达载体的宿主菌均不表达该蛋白 (图 3)。新蛋白的分子量约为78 kDa,与预期值大小一致,说明MBP-NU融合蛋白诱导表达成功。
2.3.1 诱导温度和IPTG浓度对MBP-NU融合蛋白表达量的影响
SDS-PAGE结果显示 (图4),在IPTG浓度相同的条件下,融合蛋白的相对表达量随温度的升高而增加;在温度相同的条件下,融合蛋白的相对表达量随IPTG浓度的不同出现了不同的变化,24 ℃时表达量基本相同,28 ℃时浓度为1.0 mmol/L时的表达量最大,37 ℃时浓度为0.75 mmol/L时的表达量最大。其中在37 ℃时IPTG浓度为0.75 mmol/L的条件下,融合蛋白的相对表达量最高。
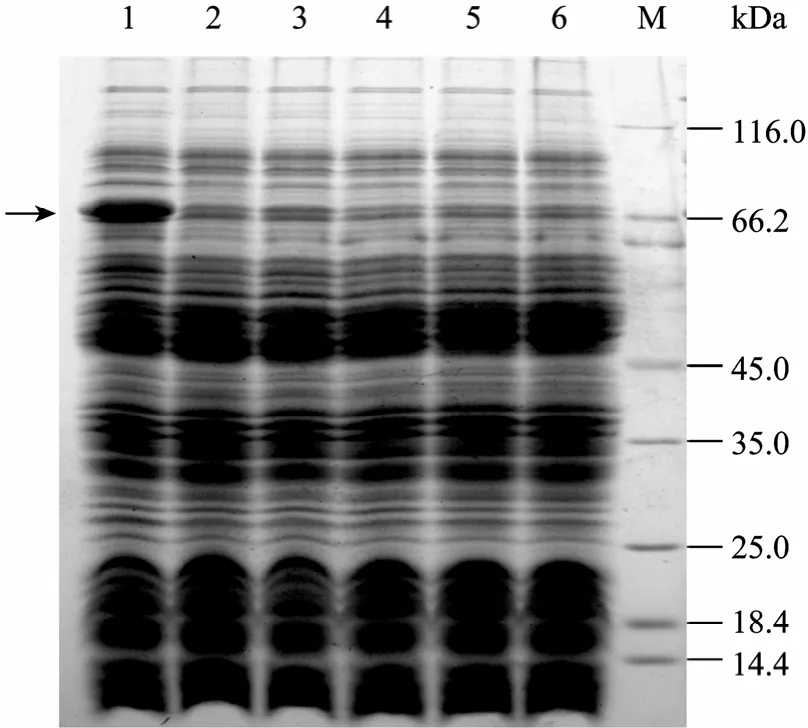
图3 MBP-NU诱导表达的SDS-PAGE分析Fig. 3 SDS-PAGE analysis of the induced expression of recombinant MBP-NU. M: protein marker; 1,3,5: induced BL21/pMAL-c4X-NU, BL21/pMAL-c4X and BL21, respectively; 2,4,6: uninduced BL21/pMAL-c4X-NU, BL21/ pMAL-c4X and BL21, respectively.
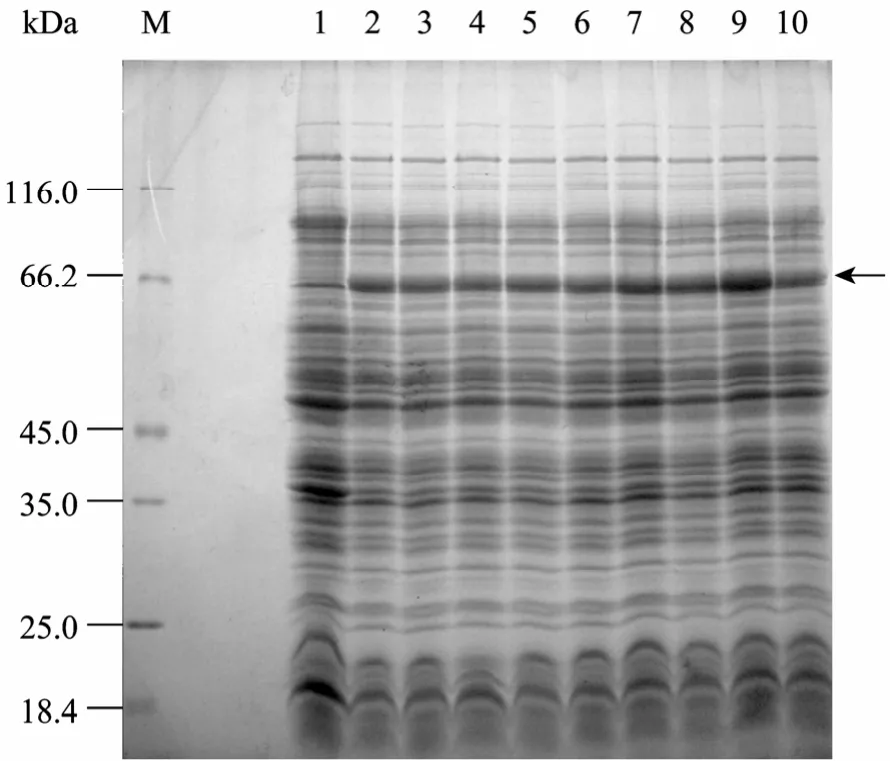
图4 不同诱导温度和IPTG浓度对MBP-NU表达量的影响Fig. 4 Effects of temperature and IPTG concentration on the expression of MBP-NU. M: protein marker; 1: uninduced; 2,3,4: induced at 24 °C; 5,6,7: induced at 28 °C; 8,9,10: induced at 37 °C; 2,5,8: induced with 0.5 mmol/L IPTG; 3,6,9: induced with 0.75 mmol/L IPTG; 4,7,10: induced with 1.0 mmol/L IPTG.
2.3.2 诱导时间对MBP-NU融合蛋白表达量的影响
SDS-PAGE结果显示 (图5),诱导时间在1.5 h时融合蛋白的相对表达量较高。诱导时间超过2.5 h,表达量有所下降。
2.4 融合蛋白的纯化
将诱导表达的菌体进行超声破碎后离心,上清液用 Amylose resin进行纯化。纯化蛋白经SDS-PAGE和 Bandscan软件分析其纯度为 93.8% (图 6),表明与 MBP融合的灵杆菌核酸酶可通过Amylose resin进行高效的纯化,从而避免了多次纯化对核酸酶稳定性及回收率的影响。
2.5 MBP-NU融合蛋白的活性检测
研究考察了不同活性浓度的 MBP-NU对DNA、RNA的水解能力,如图7所示。随着MBP-NU的浓度增加,DNA的浓度由于酶解而减少。当MBP-NU浓度达到10.0 U时,DNA已完全被降解。由图7B可知,MBP-NU可以完全降解RNA。纯化的MBP-NU的比活力为1.11×106U/mg。
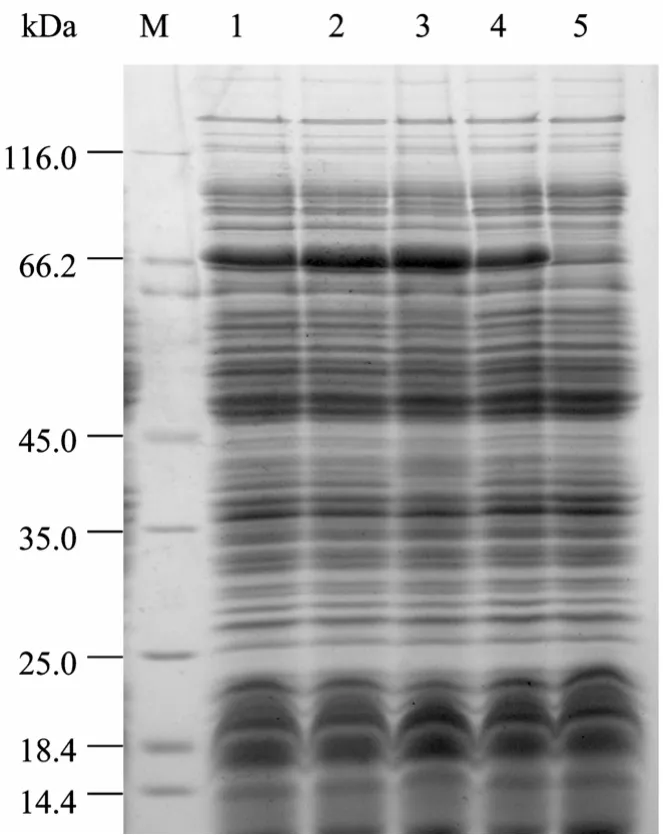
图5 不同诱导时间对MBP-NU表达量的影响Fig. 5 Effect of induction duration on the expression of MBP-NU. M: protein marker; 1−4: induced for 3.5, 2.5, 1.5, 0.5 h, respectively; 5: uninduced.
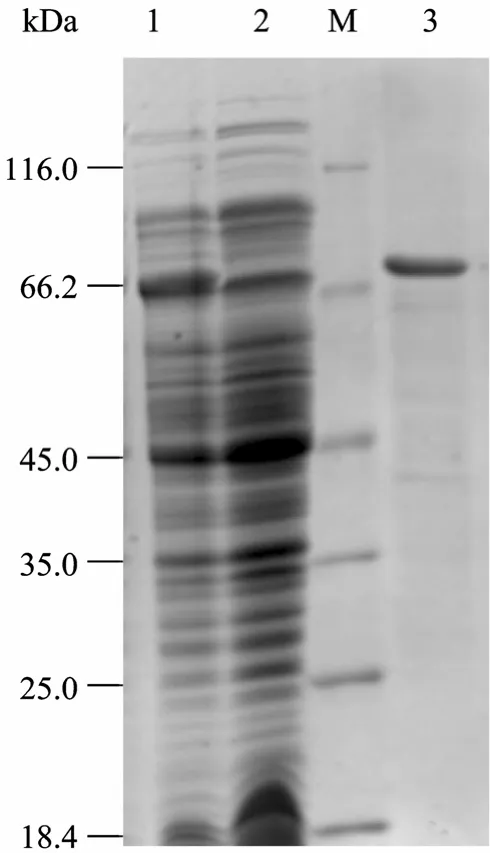
图6 纯化MBP-NU融合蛋白的SDS-PAGE分析Fig. 6 SDS-PAGE analysis of purified MBP-NU. M: protein marker; 1: induced; 2: uninduced; 3: purified MBP-NU.
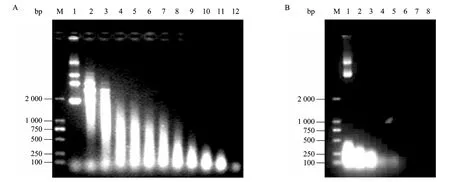
图7 MBP-NU的DNase活性 (A) 和RNase活性 (B) 检测Fig. 7 Assay of the DNase (A) and RNase (B) activity of MBP-NU. (A) 1−12: different concentration of MBP-NU(U): 0, 0.25, 0.5, 0.75, 1.0, 1.25, 1.5, 2.0, 3.0, 4.0, 5.0, 10.0. (B) 1−9: different concentration of MBP-NU (U): 0, 8, 16, 24, 32, 40, 48, 56; M: DNA marker DL2000.
2.6 EDTA和PMSF对MBP-NU活性的影响
研究考察了不同浓度的 EDTA和 PMSF对MBP-NU活性的影响,如图 8所示,0.5 mmol/L EDTA或1 mmol/L PMSF对MBP-NU的活性几乎无影响,但EDTA浓度达到1 mmol/L时,约80%的 MBP-NU活性受到抑制。
2.7 温度、pH和盐浓度对MBP-NU活性的影响
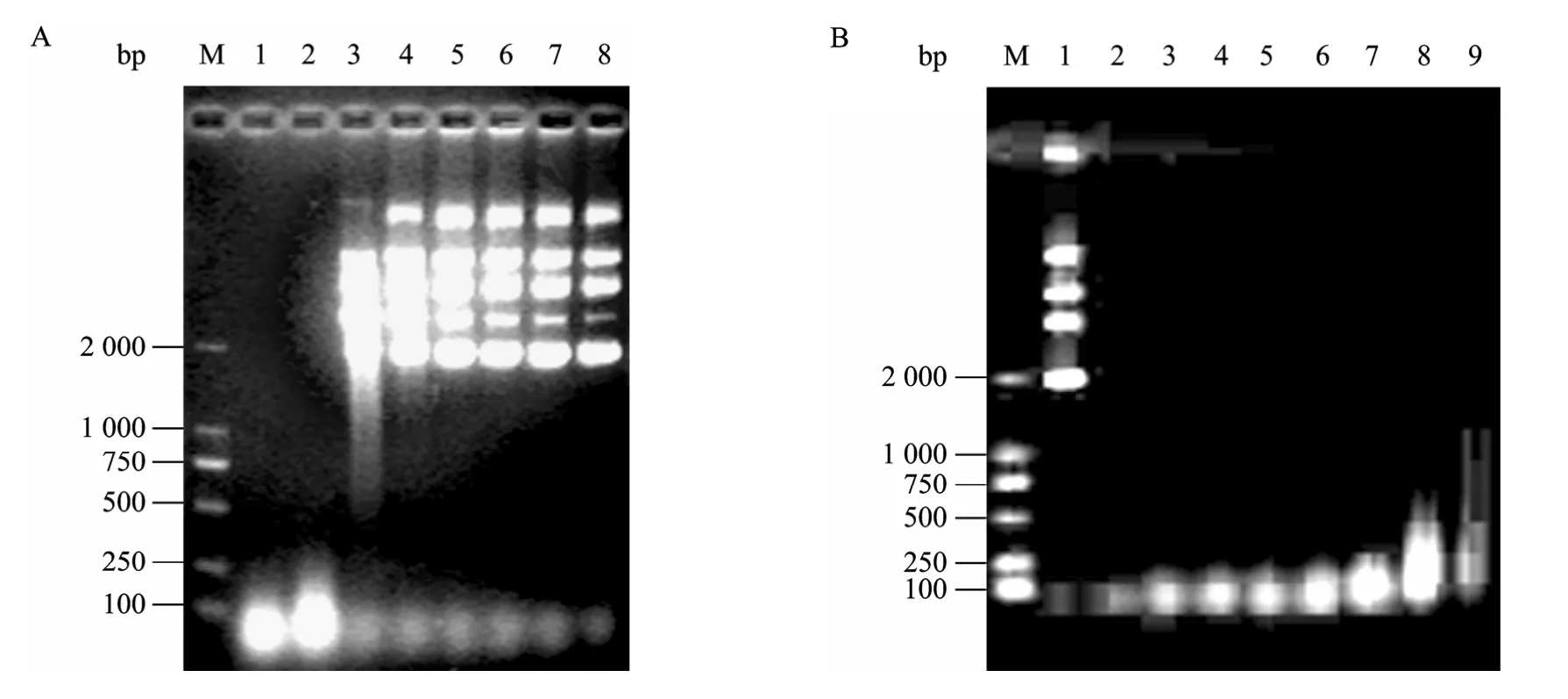
图8 EDTA (A) 和PMSF (B) 对MBP-NU活性的影响Fig. 8 Effects of EDTA (A) and PMSF (B) on the MBP-NU activity. (A) 1−7: different concentration of EDTA (mmol/L): 0, 0.5, 1.0, 2.0, 3.0, 4.0, 5.0; 8. control. (B) 1: control; 2−9: different concentration of PMSF (mmol/L): 0, 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, 12.5; M: DNA marker DL2000.
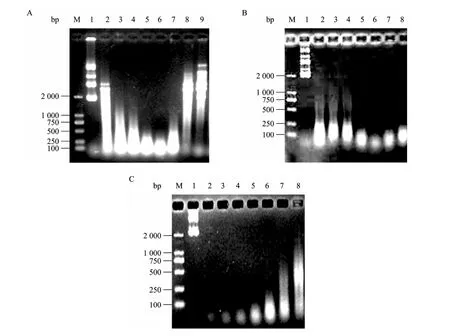
图9 温度 (A)、pH (B) 和不同浓度KCl (C) 对MBP-NU活性的影响Fig. 9 Effect of temperature (A), pH (B) and different KCl concentration (C) on the MBP-NU activity. (A) 1: control; 2−9: different temperatures (°C): 0, 4, 16, 24, 37, 52, 67, 82. (B). 1: control; 2−8: different pH: 3.7, 4.5, 5.6, 7.0, 8.0, 9.0, 10.0. (C) 1: control; 2−8: different KCl concentration (mmol/L): 0, 50, 100, 150, 200, 250, 300; M: DNA marker DL2000.
考察了不同温度、pH和盐浓度对MBP-NU活性的影响。如图9A所示,随着处理温度的升高,剩余DNA的量由于酶解而呈现先降低后增加的变化趋势,其中 37 ℃时的浓度最低,说明 MBP-NU的最适温度为37 ℃;图9B显示MBP-NU的最适pH为8.0;图9C说明,低于150 mmol/L的KCl对 MBP-NU的活性几乎没有影响,当KCl浓度为250 mmol/L时开始出现抑制现象,并随盐浓度的增加抑制程度也增加。NaCl对MBP-NU的活性影响与KCl相同 (结果未显示)。
2.8 纯化MBP-NU中蛋白酶活性的检测
以酪蛋白为底物未检测到纯化的MBP-NU中存在蛋白酶活性。分别以一定纯度的BSA和PPA为底物,与10 000 U大剂量纯化的MBP-NU长时间保温后的电泳结果显示,BSA和PPA样品的电泳带型无变化 (图 10),进一步证明本实验纯化的 MBP-NU中没有蛋白酶活性。
2.9 纯化MBP-NU与商品化Benzonase的活性比较
纯化的MBP-NU与商品化的Benzonase进行核酸的水解比较显示,在相同的酶量下,两种酶对核酸的水解效率相同 (图11)。
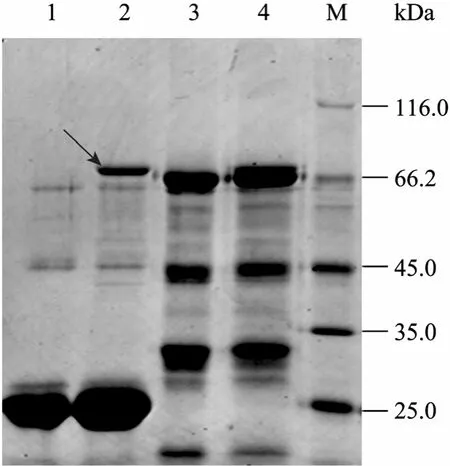
图10 纯化MBP-NU与PPA和BSA保温24 h对电泳谱带的影响分析Fig. 10 Effect of co-incubation of MBP-NU with PPA or BSA for 24 h on the SDS-PAGE bands patterns. M: protein marker; 1,2: PPA; 3,4: BSA; 1,3: control; 2,4: MBP-NU treatment. Arrows indicated the added MBP-NU.
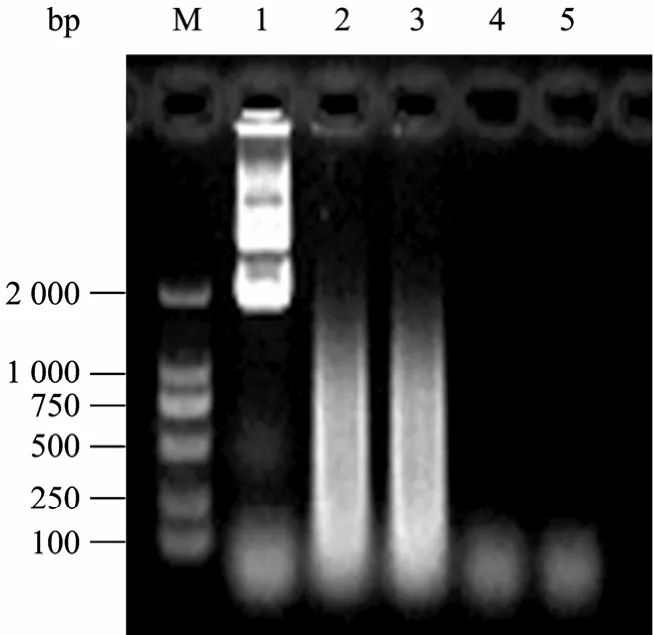
图11 纯化MBP-NU与商品化Benzonase的活性比较Fig. 11 Activity comparison between MBP-NU and Benzonase. M: DNA marker DL2000; 1: control; 2: Benzonase 1.0 U; 3: MBP-NU 1.0 U; 4: Benzonase 10 U; 5: MBP-NU 10 U.
2.10 纯化的MBP-NU除核酸效果检测
结合灵敏的PCR检测技术,采用内标法对加入的内标片段的水解程度进行检测,结果显示 (图12),伴随MBP-NU浓度的增加,目标产物的扩增量逐渐降低,当酶量超过20 U/mL时,目的条带完全消失,说明MBP-NU对于菌体裂解液中的模板有很高的水解效率。
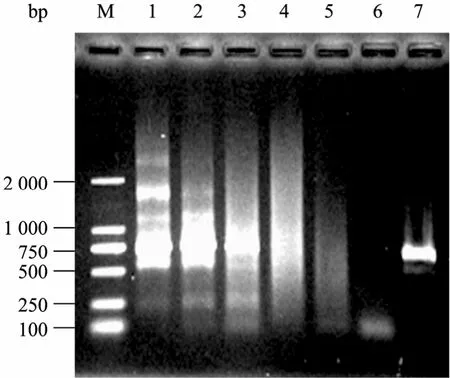
图12 不同浓度MBP-NU对菌体裂解液中内标基因NU PCR扩增的影响Fig. 12 Effect of MBP-NU treatment on the PCR amplification of internal reference gene NU in cell lysis. M: DNA marker DL2000; 1−6: different concentration of MBP-NU (U): 0, 1, 5, 10, 15, 20; 7: PCR product from 1.2.1.
3 讨论
对灵杆菌核酸酶的分泌途经、进化和作用机制已有较多的研究报道[9-12]。灵杆菌核酸酶的催化高效性使其在生物技术和实际生产中的应用价值不断凸显,特别是该酶在消除核酸污染方面的显著效果[13]。由于核酸酶本身对宿主细胞会造成强的毒害作用,从而给该酶的重组表达带来了很大困难。已有研究报道利用大肠杆菌分泌表达该酶,但由于原核分泌系统的局限性,使其表达量仅达到1.23×104U/L[6]。本研究克隆了灵杆菌的核酸酶基因,序列分析表明,其与报道的S. marcescens 核酸酶基因、Serratia proteamaculans 568的核酸酶的同源性分别为97%和85%。尝试利用分泌型原核表达载体pLLP-OmpA和pET22b表达该基因,但由于微量的胞内本底表达使菌体生长严重受抑或者死亡。
本研究构建了MBP-NU融合蛋白胞内原核表达载体,在对多个菌种进行筛选的基础上,实现了其在大肠杆菌胞内的成功表达和表达条件优化,菌体生长正常,表达量高达 1.21×107U/L。对于该酶在胞内的可溶性表达为何没有造成宿主细胞的死亡,推测原因主要有以下两个方面:一是灵杆菌核酸酶与麦芽糖结合蛋白融合表达,有助于减少其在菌体内对宿主菌的毒害;二是已有研究通过点突变的方式证明二硫键是灵杆菌核酸酶发挥催化活性所必需的[14],由于胞内的氧化还原条件抑制了二硫键的形成,使酶在胞内无水解活性,因此不影响细胞的正常生长,当细胞破碎后,外界的氧化条件促使其向有活性的形式转化。另外本研究中曾转化构建的表达载体于有利于二硫键形成的菌株 Rosetta gami2 (DE3),未有转化菌落形成,同时发现该酶仅在个别的菌株中表达,推测可能是由于不同菌株细胞内部氧化还原状态的差异使二硫键的形成存在巨大的差异,其内在的真正原因有待进一步揭示和证实。
重组酶对核酸的水解实验证明重组核酸酶在不切除MBP的情况下仍具有降解DNA和RNA的活性,最适反应条件为37 ℃、pH 8.0,蛋白纯化时缓冲液中常用的添加物0.5 mmol/L EDTA、1 mmol/L PMSF对MBP-NU的活性几乎无影响,低于150 mmol/L 的KCl或NaCl (结果未显示) 对融合蛋白MBP-NU的活性亦无影响。纯化的MBP-NU中未检测到蛋白酶活性,且该酶具有很好的储存稳定性,4 ℃条件下储存2个月酶活性几乎没有变化,其水解效率与商品化Benzonase相同。Marcin等报道细菌裂解液中添加 25 U/mL (约 25 ng/mL) 的Benzonase即可满足蛋白质纯化中核酸去除的需要,在浓度为9 U/mL时,4 h可使99%的放射性标记的核酸完全水解,可满足FDA对重组蛋白药物每次使用剂量不得超过10 pg核酸污染的需求[15]。本研究中发现表达MBP-NU菌体的裂解液粘稠度明显低于对照菌。本实验室在无机焦磷酸化酶的重组表达与纯化实验中,向含有6 g菌体的15 mL裂解液中添加终浓度为10 U/mL (约10 ng/mL) 的MBP-NU,裂解菌体的粘度在5 min内明显下降,结合PCR高灵敏度的检测极限,采用内标法证明,当酶浓度达到20 U/mL时,在5 min内即可使作为内标的DNA模板降解至PCR无法检测到的水平。以上结果说明该酶具有极高的核酸水解效率,在核酸去除中具有添加酶蛋白量少的显著特征。
本研究建立了灵杆菌核酸酶的高效胞内重组表达体系,其表达量远远高于已有的分泌重组表达系统,融合MBP的核酸酶具有很高的水解效率,这些结果为在蛋白纯化中利用酶法低成本地去除核酸奠定了基础。
REFERENCES
[1] Muro-Pastor AM, Flores E, Herrero A, et al. Identification, genetic analysis and characterization of a sugar-non-specific nuclease from the cyanobacterium Anabaena sp. PCC 7120. Mol Microbiol, 1992, 6(20): 3021−3030.
[2] Rangarajan ES, Shankar V. Sugar non-specifc endonucleases. FEMS Microbiol Rev, 2001, 25(5): 583−613.
[3] Li L, Lin S, Yanga F. Functional identification of the non-specific nuclease from white spot syndrome virus. Virology, 2005, 337(2): 399−406.
[4] Meiss G, Gast FU, Pingoud AM. The DNA/RNA non-specific Serratia nuclease prefers double-stranded A-form nucleic acids as substrates. J Mol Biol, 1999, 288(3): 377−390.
[5] Chen C, Krause K, Pettitt BM. Advantage of being a dimer for Serratia marcescens endonuclease? J Phys Chem B, 2009, 113(2): 511−521.
[6] Kim WY, Lee HS, Suh SY, et al. Purification and cellular localization of extracellular nuclease of Serratia marcescens expressed in Escherichia coli. Kor J Microbiol, 1994, 32(2): 147−154.
[7] Cattoli F, Boi C, Sorci M, et al. Adsorption of pure recombinant MBP-fusion proteins on amylose affnity membranes. J Membrane Sci, 2006, 273(1/2): 2−11.
[8] Cupp-Enyard C. Sigma’s non-specific protease activity assay-casein as a substrate. J Vis Exp, 2008, 17(19): 899.
[9] Schäfer P, Scholz SR, Gimadutdinow O, et al. Structural and functional characterization of mitochondrial EndoG, a sugar non-specifc nuclease which plays an important role during apoptosis. J Mol Biol, 2004, 338(2): 217−228.
[10] Miller MD, Cai JW, Krause KL. The active site of Serratia endonuclease contains a conserved magnesium-water cluster. J Mol Biol, 1999, 288(5): 975−987.
[11] Lunin VY, Levdikov VM, Shlyapnikov SV, et al. Three-dimensional structure of Serratia marcescens nuclease at 1.7Å resolution and mechanism of its action. FEBS Lett, 1997, 412(1): 217−222.
[12] Shlyapnikov SV, Lunin VV, Blagova EV, et al. X-ray analysis of the magnesium-containing endonuclease from Serratia marcescens. Russ J Bioorg Chem, 2001, 27(6): 370−377.
[13] Benedik MJ, Strych U. Serratia marcescens and its extracellular nuclease. FEMS Microbiol Lett, 1998, 165(1): 1−13.
[14] Ball TK, Suh Y, Benedik MJ. Disulfide bonds are required for Serratia marcescens nuclease activity. Nucl Acids Res, 1992, 20(19): 4971−4974.
[15] Olszewski M, Filipkowski P. Benzonase-possibility of practical application. Postepy Biochem, 2009, 55(1): 21−24.
猜你喜欢
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
快乐语文(2021年34期)2022-01-18
中国(俄文)(2020年8期)2020-11-23
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
天然产物研究与开发(2018年7期)2018-08-21
中国病理生理杂志(2017年2期)2017-01-17
中学化学(2016年2期)2016-05-31
