
耶尔森菌抵抗宿主免疫并诱导吞噬细胞凋亡机制研究
2011-01-24 02:12:32梁俊容综述景怀琦审校
中国人兽共患病学报 2011年8期
梁俊容(综述),景怀琦(审校)
人类先天性免疫系统在防御细菌病原体损害时依赖于巨噬细胞和多形核白细胞(嗜中性粒细胞PMNs)的协调作用。单核吞噬细胞(如单核细胞,巨噬细胞和树突状细胞)能产生炎性细胞因子,并能通过MHCⅡ类分子提呈抗原给CD4+T细胞从而通过特异性免疫清除病原体[1]。然而细菌为了能在宿主细胞中生存,诱发专职吞噬细胞凋亡对细菌而言是极为有利的[2]。
对人和啮齿动物有致病性的耶尔森菌主要分为三类——鼠疫耶尔森菌,假结核耶尔森菌和小肠结肠炎耶尔森菌。鼠疫耶尔森菌是引起鼠疫的病原体;假结核耶尔森菌则为肠系膜淋巴结炎和败血症的一种病原体;小肠结肠炎耶尔森菌在人类的感染是非常普遍的,其可以造成从急性肠炎到肠系膜淋巴结炎的一系列胃肠道症状。鼠疫耶尔森菌是经由跳蚤叮咬进入机体,而另外两者则是食源性病原体,主要通过粪口途径感染[1]。虽然这三种耶尔森菌通过不同的传播途径感染宿主,导致了严重程度不同的各种疾病,但是他们都呈现出嗜淋巴组织特性以及抵抗宿主非特异性免疫应答的能力[3]。与其它肠源性致病菌(志贺菌/沙门菌)不同,耶尔森菌主要是一种细胞外病原体。在鼠感染模型中,肠致病性耶尔森菌在肠粘膜通过M细胞侵入下层淋巴组织(即Peyer结),随后运行至淋巴结、脾、肝脏导致大量的多核白细胞增生,并与细胞外耶尔森菌形成微菌落或者微小囊肿,最终导致 Peyer结细胞结构的完全崩溃[20]。先天免疫防御系统的中性粒细胞、巨噬细胞和N K细胞以及随后发生的高效获得性免疫应答反应是克制耶尔森菌感染所必须的。特异性的抗体和产生 IFN-γ的CD4/CD8+T细胞在清除耶尔森菌的感染的过程中都起着重要的作用[4]。
1 耶尔森菌攻克宿主防御机制的Y op毒力系统及其主要的毒力因子
越来越多的证据表明:为了抵抗宿主的免疫机制,致病性耶尔森菌通过Ⅲ型分泌系统转运一系列的效应蛋白(耶尔森菌外膜蛋白 Yops)到真核细胞细胞质内来抑制宿主免疫。致病性耶尔森菌都携带一个70kb的毒力质粒p YV,这个质粒编码一套完整的蛋白摄入设备(Ⅲ型分泌系统)以及至少6种效应蛋白(Yops:Yop H,YopO/YpkA,YopP/YopJ,YopE,YopM,和 Yop T)以及他们的蛋白伴侣[4]。Zhao的实验发现:耶尔森菌p YV+菌株能诱导小鼠TNFRp55介导的感染性急性期脾细胞凋亡[5]。Ⅲ型分泌系统能够使细胞外的细菌粘附到它们的宿主细胞膜上,并插入一些孔蛋白在胞浆膜上(涉及到的蛋白质主要有LcrV,YopB,YopD),然后将 6种Yop效应子(YopE、H、T、O、P、M)传递入细胞。这些Yops的主要功能是抑制宿主的免疫应答。其中至少4种外膜蛋白 Yops(Yop H,YopE,Yop T和YopO/YpkA)通过破坏中性粒细胞以及巨噬细胞的细胞骨架来阻止巨噬细胞和多形核白细胞对耶尔森菌的吞噬作用[4]。ScottD.Mills的实验表明,耶尔森菌引起细胞凋亡需要有效的分泌系统和转运机制来将一个或多个细菌效应蛋白转运至靶细胞的细胞质中发挥作用[6]。一旦 Yops被转运至细胞质中,这些 Yop效应子将破坏细胞骨架,阻止核因子κB(nuclear factor of kappa B,NF-κB)的释放 ,抑制丝裂原活化蛋白激酶(motogen-activated protein kinase activation,MAPK)的激活,抑制细胞因子的产生并且通过诱导凋亡从而引起细胞死亡[7]。其中YopP/YopJ在诱导细胞凋亡中发挥着主要作用。
1.1 Yop H Yop H具有去磷酸化和蛋白灭活作用,是一种可以通过调节补体受体或者Fc受体来抑制吞噬作用的酪氨酸蛋白激酶[8]。它可使宿主蛋白去磷酸化,并抑制巨噬细胞和中性粒细胞调控活性氧的产生。
1.2 YopE YopE可以诱导宿主细胞肌动蛋白微丝结构的损坏从而导致宿主细胞聚集,并从细胞外基底膜上分离[21]。
1.3 Yop T我们已经从一种缺乏5种已知的 Yop效应因子的突变株(HOPEM)中证实了 Yop T的作用,Yop T是一种半胱氨酸蛋白酶,通过诱导小GTP结合蛋白 RhoA的修饰以及重分布,导致了GTP的失活[4,10]。
1.4 YopO YopO(假结核耶尔森菌和鼠疫耶尔森菌中为 YpkA)是一种能够诱导上皮细胞形态改变的丝氨酸/苏氨酸激酶[4],其与 YopE,Yop H和Yop T介导的此过程不同:他引起细胞皱缩,但不需要从细胞基底膜分离。其可能为一种细胞骨架调节的膜相关分子。
1.5 YopM YopM包含了连续的LPX重复区,是一个富亮氨酸重复超家族蛋白结合域的亚型。它和福氏志贺菌的 IpaH蛋白或鼠伤寒沙门氏菌的Ssp H1及Ssp H2蛋白同属于富亮氨酸超家族Ⅲ型效应因子。最近研究显示其可能通过破坏N K细胞来干扰先天性免疫应答[10]。
1.6 YopP(假结核耶尔森菌中称为 YopJ) 研究基因序列分析显示 YopP和 YopJ与野油菜黄单胞菌中的 AvrRxv、沙门菌中的 AvrA、根瘤菌种的y410高度相似,AvrRxv为细胞程序性死亡(凋亡)通路激活所介导过敏反应的许多无毒力蛋白质之一[6]。Monack等发现 YopP(YopJ)除了破坏吞噬细胞外还可以诱导鼠巨噬细胞的凋亡。由假结核耶尔森菌 YopJ诱导的凋亡现象已经在鼠感染实验中证实,有证据显示 YopP(YopJ)可以阻碍丝裂原活化蛋白激酶MAPK,MAPK激酶(M KKs)以及核因子(NF-κB)的激活而抑制α-肿瘤坏死因子(TNF-α)和白细胞介素IL-8的合成,并最终诱导了巨噬细胞的凋亡。同时 YopP能减少黏附因子(如内皮细胞的ICAM-1和选择素-E)的表达,抑制了趋化中性粒细胞到感染部位的能力[4]。YopP(YopJ)扰乱了信号通路的多样性,包括 NF-κB、ERK2(细胞外信号调节激酶)、JN K(c-Jun氨基末端激酶)和p38通路等丝裂原活化蛋白激酶(MAPK)的活性[9],干扰了感染靶细胞的 TNF-α,IL-1,IL-8和黑色素瘤生长刺激因子(MGSA)的合成[10]。同时 YopJ可以直接结合于MAPK激酶超家族以及NF-κB的抑制蛋白IκB,从而阻止了MAPK激酶的磷酸化以及随后的激活过程和IκB的降解[11]。
YopP/J与 IKKβ激酶和MAPK激酶之间相互作用的研究表明:YopP/J属于与类泛素蛋白(ubiquitin)蛋白酶相关的半胱氨酸蛋白酶家族成员。与类泛素蛋白蛋白酶可以清除C末端一个长度为11kDa的小泛素相关因子SUMO-1。YopJ催化172位半胱氨酸功能的突变导致其蛋白酶丧失活性,从而阻碍了这些外膜蛋白对NF-κB和M KK抑制信号的传导以及诱导细胞凋亡的功能。已证实172位的半胱氨酸残基和143位的精氨酸残基仅仅存在于高毒力的小肠结肠炎耶尔森菌及鼠疫和假结核耶尔森菌的 YopJ中。
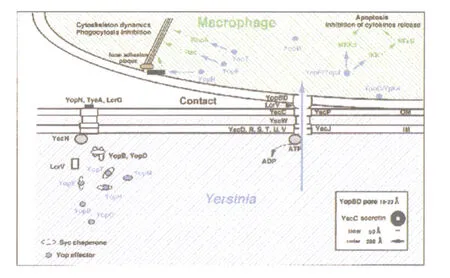
图1 耶尔森菌与巨噬细胞相互作用假设模型[10]
2 耶尔森菌与细胞(巨噬细胞/中性粒细胞/树突细胞)的相互作用
当耶尔森菌在37℃营养丰富的环境中生存时,Ysc分泌系统被激活,合成并储存 Yop蛋白。这些蛋白中,有些结合特定的SYC蛋白伴侣帽盖(可能是防止蛋白不成熟时的缔合)。只要耶尔森菌未与真核细胞接触,由 YopN-TyeA-LcrG组成的 YSC分泌通道控制阀门处于关闭状态。一旦缺Ca2+或与真核细胞接触后,分泌通道的阀门打开,易位子YopB在 YopD和LcrV的帮助下嵌入真核细胞。Yop效应子(Yop H,YopO/YpkA,YopP/YopJ,YopE,YopM和 Yop T)随后通过分泌通道转运,在易位子的控制下到达靶细胞的细胞质内发挥作用[10]。其中 YopE和 Yop T作用于细胞骨架,而YopP/YopJ诱导细胞凋亡(图1)。
2.1 Ysc分泌系统编码分泌系统的基因位于耶尔森菌毒力质粒p YV的 4个连续基因座上:virA(yopN,tyeA,sycN,yscX、Y、V、lcrR),virB(yscN-U),yscW以及virC(yscA-M)。大多数的Ysc蛋白对于分泌过程是必需的,其中有4种蛋白(YscD,YscR,YscU和 YscV)跨越耶尔森菌内膜。
2.2 伴侣蛋白 Yop效应蛋白和转运蛋白的分泌需要其对应的蛋白伴侣(Syc)。通过与 Yops结合,Syc(蛋白伴侣)可以保证蛋白质的稳定性,保持适合的构象。在分泌时,蛋白伴侣从伴侣 Yop上释放,转运结构域可以自由与转运装置结合,然后转运装置引导 Yops进入宿主细胞胞浆[10]。
2.3 分泌信号 Yops需要在特殊的靶点被分泌系统识别后才能分泌。现发现Ⅲ型分泌系统有两种不同的分泌信号控制 Yops的分泌:(1)N末端(或5′mRNA)分泌信号。(2)伴侣蛋白介导的信号[11]。
2.4 跨膜转运 迄今为止,已知有6个效应蛋白(Yop H,YopO/YpkA,YopP/YopJ,YopE,YopM和Yop T)通过一个定向的过程穿过真核细胞膜转运。这同时需要 YopB/YopD的参与。在12种分泌蛋白中,只有 YopB/YopD具有疏水结构域,说明它们可以与膜相互作用,这两种蛋白的分泌及组装可能还需要LcrV协助[10]。
3 耶尔森菌引起巨噬细胞的凋亡
耶尔森菌和巨噬细胞的相互作用最终激活了巨噬细胞本身的死亡程序。凋亡在一些感染性疾病中发挥了重要作用。耶尔森菌能诱导体外培养的巨噬细胞凋亡,表现为膜性结构形成囊泡(凋亡体形成)、胞浆皱缩、DNA片段化等凋亡的普遍特征[7]。其诱导吞噬细胞凋亡的机制主要如下。
3.1 抑制巨噬细胞 TNF-α的产生α-肿瘤坏死因子(TNF-α)主要是由巨噬细胞分泌的一种致炎细胞因子,对于减少耶尔森菌感染的严重性方面至关重要。相应的,抑制 TNF-α的合成增强了耶尔森菌在宿主中增殖的能力。脂多糖(LPS)激活 ERK1/2、JU K和p38通路等丝裂原活化蛋白激酶(MAPK),被活化的MAPK激活NF-κB使得 TNF-α开始转录,而后刺激巨噬细胞和中性粒细胞PMN杀灭微生物。然而耶尔森菌与巨噬细胞相互作用释放 YopP/YopJ可以通过激活细胞凋亡蛋白酶(Caspase)来诱导细胞凋亡。同时 YopP/YopJ对MAPK进行反向调节并影响NF-κB的激活,这两种作用可以解释YopP/YopJ引起的 TNF-α减少。
3.2 耶尔森菌引起多形核白细胞产生活性氧分子ROS(Reactive Oxygen Species) 多形核白细胞的凋亡与ROS产量增加密切相关。在有 TNF-α和细菌存在时,由慢性肉芽肿(CGD)病人产生的多形核白细胞或者使用还原型辅酶Ⅱ(NADPH)抑制剂处理的多形核白细胞可以延迟凋亡的产生。进一步的研究证实,使用可以诱导产生活性氧分子的佛波脂(phorbolmyristate acetate)处理多形核白细胞后会加速凋亡进程;且ROS水平的升高会抑制多形核白细胞凋亡蛋白酶的活性[1]。多形核白细胞感染耶尔森菌后,毒力质粒型的耶尔森菌株抑制多形核白细胞ROS的产生与磷脂酰丝氨酸暴露不足、乳酸脱氢酶(LDH)释放、EthD-1着色有关。我们发现在多形核白细胞与鼠疫耶尔森菌吞噬作用中,使用NADPH氧化酶抑制剂抑制中性粒细胞ROS的产生可以降低LDH释放和 EthD-1着色[13]。由此可以看出ROS在一定程度上影响多形核白细胞的凋亡,耶尔森菌依靠 TTSS抑制ROS的产生从而延缓正常多形核白细胞的凋亡[1]。
3.3 耶尔森菌对巨噬细胞内信号转导途径的作用
3.3.1 抑制转录因子NF-κB的激活 转录因子NF-κB是先天性免疫的核心,它控制细胞因子、急性期蛋白、粘附分子的合成,并通过抑制细胞凋亡来调节细胞生存[12]。Mittal的实验结果显示耶尔森菌抑制NF-κB的激活与触发巨噬细胞凋亡并抑制α-肿瘤坏死因子(TNF-α)的产生之间存在着密切联系。并且O∶8血清型耶尔森菌的毒力蛋白 YopP比O∶9血清型的 YopP更能有效地抑制NF-κB从而诱导巨噬细胞凋亡。目前认为耶尔森菌作用于巨噬细胞NF-κB信号途径的过程是 YopP/J结合并抑制了巨噬细胞的NF-κB的活化激酶IKKβ。游离的NF-κB被转运到细胞核中并结合在靶序列和转运启动子上。YopP所诱导的细胞凋亡可被一时性过度表达的 NF-κBp65特异地抑制,表明 YopP介导的细胞凋亡是由于 NF-κB信号途径遭到破坏所致[12]。
暴露于细菌脂多糖(LPS)或者其他炎性刺激因子例如α-肿瘤坏死因子(TNF-α)或者白介素1(IL-1)、基因诱变剂和电离辐射等可激活巨噬细胞NF-κB途径[3]。NF-κB抗细胞凋亡是一个涉及多个信号通路的复杂过程,但其主要方式是通过诱导或上调抗凋亡基因的表达实现的。这些基因调节位点上有NF-κB的结合位点,他们的表达产物通过抑制细胞凋亡的死亡受体途径或线粒体途径发挥作用。到目前为止,研究发现细胞凋亡抑制蛋白(inhibitor of apoptosis proteins,IAPs)、Bcl-2 家族、TNFR-associated factor(TRAF1,TRAF-2)、JN K、c-FLIP、IEX-1L等都参与 NF-κB激活后的抗细胞凋亡过程。
3.3.2 促进Caspase的激活 凋亡中最早并且最常见的现象就是启动Caspase的激活。Caspase抑制剂Zvad-fmk并不阻碍耶尔森菌诱导的凋亡,这提示耶尔森菌可能在 Caspase上游水平起始凋亡[14]。Caspase通过与受体蛋白的相互作用或者清除细胞内级联反应中上游蛋白酶诱导的蛋白质水解来活化。目前的研究揭示引起凋亡主要有两条途径:细胞外的死亡受体途径及细胞内的线粒体途径。前者主要是刺激因素激活细胞内 P53,Fas,Bcl-2,NF-κB等基因,由肿瘤坏死因子(TNF)受体家族介导,使Caspase-8激活;后者是通过刺激因素影响细胞内线粒体的膜电位,导致线粒体内细胞色素C释放,胞内 Ca2+浓度升高,p H值下降,使 Caspase-9激活。两个途径最后都导致效应性Caspase-3活化,效应性 Caspase-3,-6,-7激活内切核酸酶,使DNA链断裂,最终细胞结构的全面解体[22]。环境因素、衰老和程序发展(包括从线粒体内膜释放细胞色素C至细胞质中导致的蛋白激酶自身活化)都可以引起凋亡小体复合物的聚集和激活从而激发Caspase级联反应。然而诱发细胞色素C释放的机制目前还不清楚。在 Guy R.Cornelis的试验中显示,小肠结肠炎耶尔森菌中Caspase的激活在 YopP介导的细胞死亡中起了关键作用。并且 YopP介导的细胞凋亡启动了细胞死亡通路上游的细胞溶质蛋白Bid。Bid的清除随后导致了细胞色素C从线粒体中释放出来,最终导致了 Caspase-9和Caspase-3/7的激活[14]。
3.3.3 阻止MAPK信号转导途径一系列研究显示耶尔森菌通过下调丝裂原活化蛋白激酶(MAPK)的活性来阻止巨噬细胞产生α-肿瘤坏死因子(TNF-α)。Orth的实验表明,YopP/YopJ结合并且失活MAPK上游启动子——MAPK激酶超家族的成员。革兰氏阴性菌的脂多糖能够激活巨噬细胞三种不同的MPA K家族:ERK,JN K/SAPK和p38。然而脂多糖激活MAPK的机制目前还不明确[15]。用 MAPK和 TNF-α抑制剂处理后能诱导树突状细胞凋亡。相反,在巨噬细胞中抑制p38,JN K和MEK1/2等丝裂原活化蛋白激酶并不能恢复YopP突变株诱导细胞死亡的能力,这与先前在巨噬细胞J774A.1的研究发现一致:抑制MAPK的活性从而引起耶尔森菌诱导的细胞死亡仅仅在NF-κB也被抑制的情况下才会发生[16]。
Bliska等研究发现,假结核耶尔森菌YopJ促进了丝氨酸/苏氨酸的乙酰化,从而抑制了如 MKK6等MKKs的活性,由此介导了对 NF-κB信号转导通路的抑制[17]。目前的研究发现,YopP促进了死亡诱导信号复合体(DISC)的形成,从而导致了Caspase-8的激活[14]。因此,我们推测 YopP可能在DISC水平上通过乙酰化MKK来促进Caspase-8的激活。在Autenrieth的实验中通过使用MAPK抑制剂证实:耶尔森菌毒力蛋白 YopP通过阻止宿主细胞MAPK信号转导途径引起OVA摄取的减少,从而影响了树突状细胞诱导产生 T淋巴细胞的能力[18]。
3.4 脂多糖对吞噬细胞凋亡的促进作用 我们分析了YopP在诱导巨噬细胞和树突细胞凋亡过程中的区别。在树突细胞中,脂多糖在耶尔森菌感染早期阶段通过清除Caspase-8从而在 YopP诱导凋亡的过程中发挥了重要作用[14]。在J774A-1巨噬细胞中,脂多糖通过抑制 YopP介导的NF-κB的激活来促进凋亡。Bohn的实验提出耶尔森菌脂多糖可刺激小鼠腹腔巨噬细胞产生IL-18和 IL-12。感染初期IL-18可负反馈抑制 IL-12过度产生,IL-18、IL-12共同诱导N K细胞或者CD4+T细胞分泌IFN-γ后,IFN-γ又会诱导巨噬细胞产生 IL-12,导致级联放大效应,生成高水平 IFN-γ,达到最佳保护机制[19]。
4 存在的问题及今后研究方向
总之,在耶尔森菌抵抗宿主免疫和诱导细胞凋亡过程中,许多蛋白因子参与了其信号转导。尽管耶尔森菌引起细胞凋亡在体外实验中是明显的,但其生理病理学作用目前并不清楚。YopP/YopJ突变不影响毒力,至少在小鼠模型上是这样。用 Yop突变株逐一筛选的实验结果表明 Yop能诱发巨噬细胞凋亡,而与 YopE无关,其确切机制仍有待探讨[2]。此外,耶尔森菌诱导的细胞凋亡从未在体内试验中观察到。唯一知道的是,小肠结肠炎耶尔森菌感染小鼠,受染的 Peyer结上凋亡细胞数增加。这种现象是因为感染组织的一般退化还是由于YopP/YopJ的作用,依然有待证明。虽然 YopP/YopJ能快速引起鼠巨噬细胞和树突状细胞凋亡,然而他们对于中性粒细胞的作用仍不十分清楚。与巨噬细胞不同的是,其他细胞(如成纤维细胞和上皮HeLa细胞)在感染耶尔森菌后,尽管 YopP/YopJ被有效转运至 HeLa细胞并抑制了NF-κB的激活和IL-8细胞因子的产生,但其并不引起凋亡[16]。对耶尔森菌抵抗宿主免疫及诱导凋亡的相关信号转导通路具体机制的研究,将有助于加深对耶尔森菌引起疾病发生的分子机制的认识,也可为治疗这些疾病开拓新的途径。
[1]Spinner JL,Seo KS,O’Loughlin JL,et al.Neutrophils are resistant to Yersinia YopJ/P-induced apoptosis and are protected from ROS-mediated cell death by the type III secretion system[J].PLoS One,2010,5(2):9279.
[2]Ruckdeschel K,Roggenkamp,Lafont V,et al.Interaction ofYersinia enterocoliticawith macrophages leads to macrophage cell death through apoptosis[J].Infect Immun,1997,65(11):4813-4821.
[3]Ruckdeschel K,Harb S,Roggenkamp A,et al.Yersinia enterocolitica impairs activation of transcription factor NF-kappaB:involvement in the induction of programmed cell death and in the suppression of the macrophage tumor necrosis factor alpha production[J].J Exp Med,1998,187(7):1069-1079.
[4]Trulzsch K,Geginat G,Sporleder T,et al.Yersinia outer protein P inhibits CD8 T cell priming in the mouse infection model[J].J Immunol,2005,174(7):4244-4251.
[5]Zhao YX,Lajoie G,Zhang H,et al.Tumor necrosis factor receptor p55-deficient mice respond to acute Yersinia enterocolitica infection with less apoptosis and more effective host resistance[J].Infect Immun,2000,68(3):1243-1251.
[6]Mills SD,Boland A,Sory MP,et al.Yersinia enterocolitica induces apoptosis in macrophages by a process requiring functional type III secretion and translocation mechanisms and involving YopP,presumably acting as an effector protein[J].Proc Natl Acad Sci U S A,1997,94(23):12638-12643.
[7]Pandey AK,A Sodhi.Recombinant YopJ induces apoptosis in murine peritoneal macrophages in vitro:involvement of mitochondrial death pathway[J].IntImmunol,2009,21(11):1239-1249.
[8]Ruckdeschel K,Roggenkamp A,Schubert S,et al.Differential contribution of Yersinia enterocolitica virulence factors to evasion of microbicidal action of neutrophils[J].Infect Immun,1996,64(3):724-733.
[9]Ruckdeschel K,Machold J,Roggenkamp A,et al.Yersinia enterocolitica promotes deactivation of macrophage mitogen-activated protein kinases extracellularsignal-regulated kinase-1/2,p38,and c-Jun NH2-terminal kinase.Correlation with its inhibitory effect on tumor necrosis factor-alpha production[J].J Biol Chem,1997,272(25):15920-15927.
[10]Bleves S,GR Cornelis.How to survive in the host:the Yersinia lesson[J].Microbes Infect,2000,2(12):1451-1460.
[11]Fallman M,Persson C,Schesser K,et al.Bidirectional signaling between Yersinia and its target cell.Folia Microbiol(Praha)[J],1998,43(3):263-273.
[12]Ruckdeschel K,Mannel O,Richter K,et al.Yersinia outer protein P of Yersinia enterocolitica simultaneously blocks the nuclear factor-kappa B pathway and exploits lipopolysaccharide signaling to trigger apoptosis in macrophages[J].J Immunol,2001,166(3):1823-1831.
[13]Brodsky IE,R Medzhitov.Reduced secretion ofYopJby Yersinia limits in vivo cell death but enhances bacterial virulence[J].PLoS Pathog,2008,4(5):e1000067.
[14]Grobner S,Autenrieth SE,Soldanova I,et al.Yersinia YopP-induced apoptotic cell death in murine dendritic cells is partially independent from action of caspases and exhibits necrosis-like features[J].Apoptosis,2006,11(11):1959-1968.
[15]Orth K,Palmer LE,Bao ZQ,et al.Inhibition of the mitogenactivated protein kinase kinase superfamily by a Yersinia effector[J].Science,1999,285(5435):1920-1923.
[16]Ricote M,Garcia-Tunon I,Fraile B,et al.P38 MAPKprotects against TNF-alpha-provoked apoptosis in LNCaP prostatic cancer cells[J].Apoptosis,2006,11(11):1969-1975.
[17]Bliska JB.Yersinia inhibits host signaling by acetylating MAPK kinases[J].ACS Chem Biol,2006,1(6):349-351.
[18]Autenrieth S,Soldanova I,Rosemann R,et al.Yersinia enterocolitica YopP inhibits MAP kinase-mediated antigen uptake in dendritic cells[J].Cell Microbiol,2007,9(2):425-437.
[19]Bohn E,Sing A,Zumbihl R,et al.IL-18(IFN-gamma-inducing factor)regulates early cytokine production in,and promotes resolution of,bacterial infection in mice[J].J Immunol,1998,160(1):299-307.
[20]景怀琦,徐建国.小肠结肠炎耶尔森菌感染性疾病[J].疾病监测,2005,20(09):449-450.
[21]李振军,邱海燕,王鑫,景怀琦.O∶9血清型小肠结肠炎耶尔森菌感染病理学特征研究[J].中国人兽共患病学报,2008,24(5):418-420.
[22]Jaattela M,Tschopp J.Caspase-independent cell death in T lymphocytes[J].Nat Immunol 2003,4:416-423.
猜你喜欢
中国人兽共患病学报(2023年8期)2023-09-18 02:53:38
科学(2020年2期)2020-08-24 07:57:00
植物保护(2019年2期)2019-07-23 08:40:58
中国人兽共患病学报(2019年6期)2019-07-16 08:24:40
饲料博览(2019年1期)2019-02-14 21:44:22
动物医学进展(2017年8期)2017-10-11 08:23:32
兽医导刊(2016年12期)2016-05-17 03:51:53
吉林大学学报(医学版)(2015年1期)2015-12-17 07:47:17
医学研究杂志(2015年3期)2015-06-10 06:41:52
大理大学学报(2012年12期)2012-03-29 00:48:52
