
OⅡ空位BaZrO3晶体的第一性原理研究
2011-01-04 02:07宋兆涛陈天池邓小兵
天津师范大学学报(自然科学版) 2011年1期
宋兆涛,赵 辉,2,陈天池,邓小兵
(1.天津师范大学 物理与电子信息学院,天津 300387;2.鞍山师范学院 物理系,辽宁 鞍山 114005)
OⅡ空位BaZrO3晶体的第一性原理研究
宋兆涛1,赵 辉1,2,陈天池1,邓小兵1
(1.天津师范大学 物理与电子信息学院,天津 300387;2.鞍山师范学院 物理系,辽宁 鞍山 114005)
利用第一性原理对BaZrO3块材的OⅡ空位进行研究.由于存在OⅡ空位,O原子分别与Zr原子和Ba原子发生相互作用,原子弛豫改变晶体的结构,从而使晶常数格a,b和c各不相同.因此,通过对布居数、态密度和能带进行分析,证明OⅡ空位使BaZrO3晶体结构发生了改变.
第一性原理;BaZrO3晶体;OⅡ空位;布居数;态密度;能带
近年来,铁电体[1-4]的理论研究工具不断更新,第一性原理(First Principles)计算的兴起和应用就是其中一个重要方面.铁电体从发现到现在已经有90多年的历史了,但是如何探索新型的高性能铁电材料这一问题却依旧困扰着人们.寻找新型材料的传统方法不但需要消耗大量的人力物力,而且效率非常低.随着现代计算机技术的飞速发展,使用现代计算技术,利用第一性原理从分子原子的层面来计算材料的物理性能已成为可能.
锆酸钡(BaZrO3)是一种典型的钙钛矿型晶体,具有高熔点、低热膨胀系数和低介电损耗等特殊的物理性质.山东大学的张超[5]等对BaZrO3(001)表面进行研究,计算了晶体的结构、态密度、能带和表面能.结果表明:与PbZrO3相比,BaZrO3表面的褶皱相对较小,表面能较大,因此BaZrO3结构比较稳定,表面难以构造.
本研究从理论计算方面,利用Material studio 3.1中的Castep软件包,通过对OⅡ空位BaZrO3晶体的原子弛豫[6-8]及其相互作用进行讨论,得到OⅡ空位对BaZrO3结构造成的影响.
1 计算方法和模型
研究中的计算由 Material studio 3.1中的Castep软件包完成.Castep软件是一个基于密度泛函方法的从头算量子力学程序.它利用总能量平面波赝势方法,将离子势用赝势替代,电子波函数通过平面波基组展开,电子相互作用的交换和相关势由局域密度近似(LDA)或广义梯度近似(GGA)进行校正,这是目前较为准确的电子结构计算的理论方法.
密度泛函理论中,单电子运动的薛定谔方程可以表示为(原子单位):

在模拟过程中,采用周期性边界条件,单电子轨道波函数满足布洛赫(Bloch)定理,可采用平面波基组展开为

式(2)中,g是原胞的倒格矢,k是第一布里渊区的波矢,cki(g)是单电子轨道波函数的傅里叶(Fourier)级数.
本研究利用第一性原理密度泛函理论[8-9]对BaZrO3晶体进行计算,采用广义梯度近似[8-10]下的平面波超软赝势方法进行研究.平面波截止能量为380eV,k点网格大小为6×6×6,结构优化的总能收敛精度为1.0×10-5eV/atom.BaZrO3的单胞结构属于PM-3M空间点群,立方相的晶格常数为a=b=c=0.419 1nm,体积V=7.363 1×10-2nm3,如图1所示.
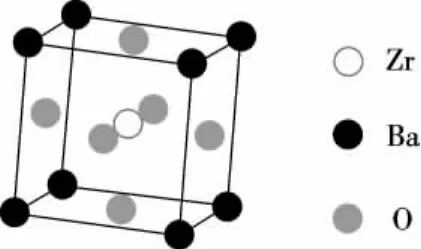
图1 BaZrO3单胞Figure 1 Unit cell of BaZrO3
2 计算结果
2.1 理想BaZrO3晶体超胞
理想BaZrO3晶体超胞(2×2×1)的空间点群为PM-3M,具有立方对称性的晶体结构,如图2所示.
2.1.1 电子弛豫和布居数
表1是理想BaZrO3超胞经Castep软件优化后各原子的坐标.几何优化能够自动根据原子的受力情况来调整原子的位置,直到所有原子的受力都为0,达到给定条件下的最稳定结构.由表1中可以看出,在分数坐标中,优化前后各原子的相对位置没有变化.
椎管内神经鞘瘤是一种起源于神经鞘膜雪旺细胞的良性肿瘤,多为单发,是椎管内髓外硬膜下常见的肿瘤之一[1-2],椎管内神经鞘瘤因其生长部位不同具有不同特征,目前关于椎管内表现为神经出入征的神经鞘瘤影像表现报道较少。现报道我院具有神经进出征的椎管内神经鞘瘤1例。

图2 理想BaZrO3晶体超胞Figure 2 Perfect crystal supercell of BaZrO3

表1 理想BaZrO3超胞优化后各原子的坐标Table 1 Atomic coordinates of geometry optimization of perfect BaZrO3crystal supercell
表2是理想BaZrO3超胞优化前后的晶格常数和体积的变化.由表2可以得出,与优化前相比,优化后的晶格常数和体积均有所增加,其中晶格常数a,b和c增加了0.43%,相应的晶胞体积增加了1.3%,平均差值在1.0%左右,差值很小.

表2 理想BaZrO3超胞优化前后晶格常数和体积的变化Table 2 Lattice constants and volume changes before and after optimization of perfect BaZrO3 crystal supercell
经Castep软件计算,理想BaZrO3超胞中各原子轨道上电子布居数和键布居数如表3所示.由表3可知,Zr原子优化前的电子构型为4s24p64d25s2,优化后为(4,5)s2.354p5.764d2.02,可见优化后Zr原子s轨道和4p轨道的电子数分别减少1.65和0.24,而4d轨道增加0.02,总电子数减少1.87.键布居中可看到Zr原子与O原子形成的共价键键长约为0.210nm,二者距离很近,所以电子轨道存在一定重叠.Zr原子的4p轨道和4d轨道得到少量电子,s轨道失去1.65e,而4s轨道电子由于定域性极强,几乎不参与成键,所以Zr原子5s轨道和O原子2p轨道有很强的重叠,形成共价键,Zr原子为电子的施主.

表3 理想BaZrO3超胞中各原子轨道电子布居数和键布居数Table 3 Atomic and bond populations on atomic orbits of perfect BaZrO3crystal
Ba原 子 优 化 后 的 电 子 构 型 为 6s1.95p6.01d0.66,与优化前的电子构型6s25p64d2相比,优化后的Ba原子6s和5p轨道电子数相差不大,4d轨道电子数减少1.34,总电子数减少1.38.从键布居中可看到Ba原子与O原子的键长大约为0.298nm,原子距离很远.Ba原子的6s和5p轨道电子定域性很强,几乎不参与成键,而Ba原子4d轨道与O原子之间相互作用,所以Ba原子和O原子形成很强的离子键.
优化前O原子的电子构型为2s22p4,优化后为2s1.952p5.13.所以优化后的 O 原子2s轨道定域性强,其电子数减少0.05;而2p轨道电子数增加1.13,总电子数增加1.08.O与O之间的相互作用很弱,布居数为-0.18e,O原子为电子的受主.
综上所述,O原子与Ba原子,O原子与Zr原子之间具有很强的相互作用.
2.1.2 态密度及分态密度分析
BaZrO3晶胞中各原子的相互作用使得晶胞中部分电子共有化,在晶胞中各原子的外层电子和价电子为整个晶胞所共有,所谓态密度也就是共有化电子在不同能级的分布图,图3是理想BaZrO3晶体的态密度及分态密度图.
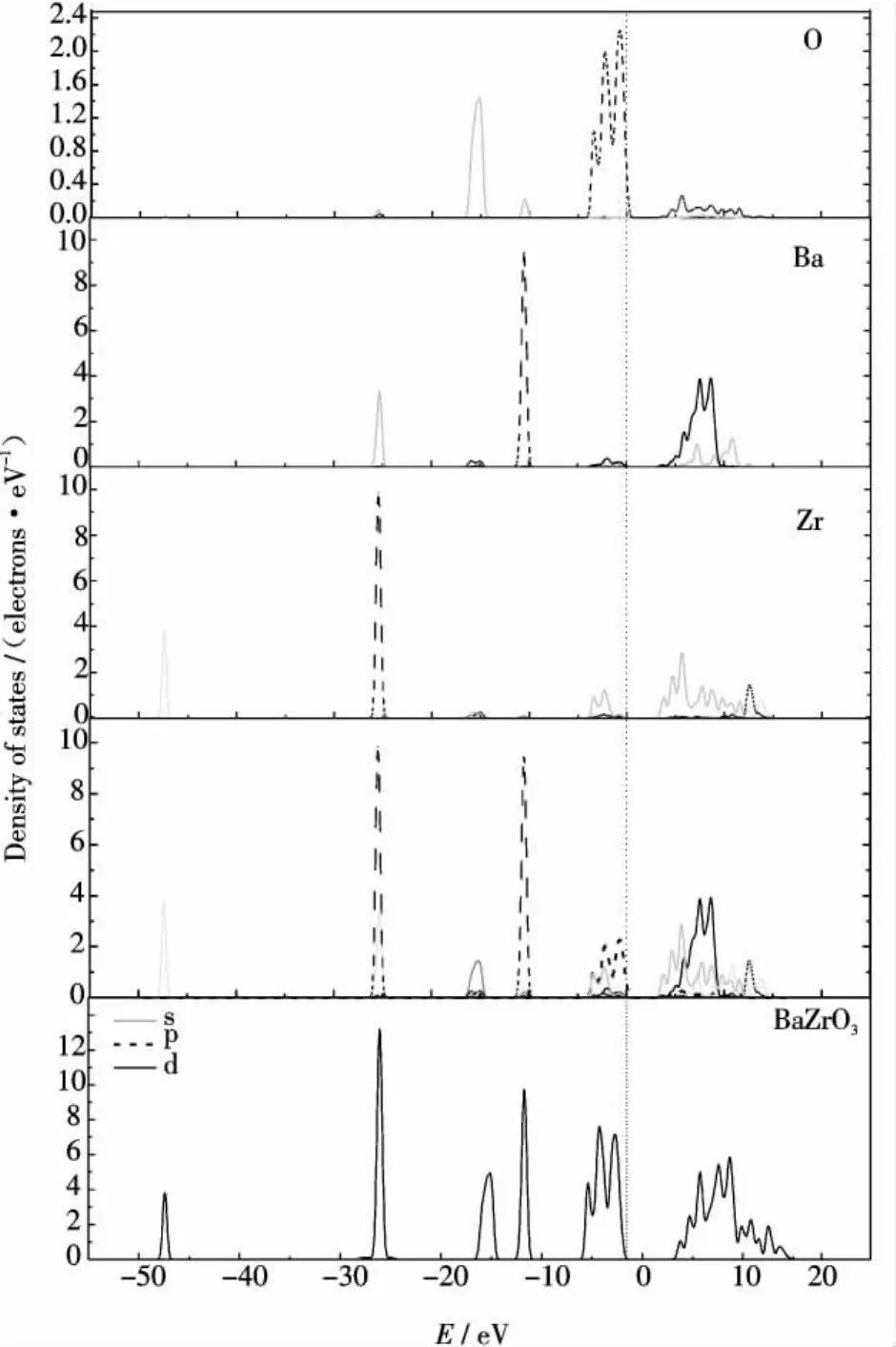
图3 理想BaZrO3晶体的态密度及分态密度Figure 3 DOS and PDOS of perfect BaZrO3crystal
由图3中可以看出,BaZrO3在-47eV处的态密度峰主要由Zr原子的4s轨道电子贡献,-25eV处的态密度峰主要由Ba原子的5s轨道电子和Zr原子的4p轨道电子贡献,-15eV处的态密度峰主要由O原子2s轨道电子贡献,-10eV处主要由Ba原子5p轨道内层电子贡献,而Zr原子4s轨道和4p轨道、Ba原子5s轨道和5p轨道以及O原子2s轨道均为内层电子,因此,以上位置的态密度峰峰形尖锐,电子的定域性很强,几乎不成键.-4~1eV之间的价带主要由O原子2p轨道电子构成,带宽为5.23eV,同时也有一部分由Zr原子4d轨道电子贡献.1eV以上的导带部分主要由Ba原子的4d和6s轨道以及Zr原子的4d轨道电子构成,同时也有一部分由O原子2p轨道电子贡献,带宽为12.61eV.可见O原子2p轨道、Ba原子6s轨道和4d轨道及Zr原子4d轨道在导带和价带区域都存在明显杂化,这说明BaZrO3晶体中O原子分别与Ba原子和Zr原子存在着强烈的相互作用.
2.1.3 能带结构
图4是计算得到的理想BaZrO3晶体的能带结构图.其中,0eV处为费米能级,费米能级以上是导带,费米能级以下是价带.BaZrO3晶体价带的顶部和导带的底部在同一k点处,由此可以判断BaZrO3晶体为直接能隙晶体.导带带宽较大说明它们参与了与O原子2p轨道成键.通过计算可以得到价带与导带之间能隙为3.23eV.

图4 理想BaZrO3晶体能带图Figure 4 Band structure of perfect BaZrO3crystal
2.2 OⅡ空位BaZrO3晶体超胞
在BaZrO3晶胞中,从xOy界面去掉ZrO2面中的1个氧原子,即为OⅡ空位,如图5所示.然后,经过对称性操作和几何优化,晶体的结构由立方相(PM-3M)转变为正交结构(PMMM).

图5 OⅡ空位BaZrO3超胞Figure 5 BaZrO3crystal supercell of OⅡvacancy
2.2.1 电子弛豫及布居数
经过几何优化,OⅡ空位BaZrO3超胞各原子坐标如表4所示.由表4中可以看出,离氧空位较近的O原子向远离氧空位的方向运动,而其余O原子会向氧空位弛豫.离氧空位较近的2个Zr原子会沿着y轴远离氧空位,而离氧空位较远的2个Zr原子会沿着y轴向氧空位弛豫.所有Ba原子均沿着x轴方向远离氧空位.因此,BaZrO3晶格参数发生变化.
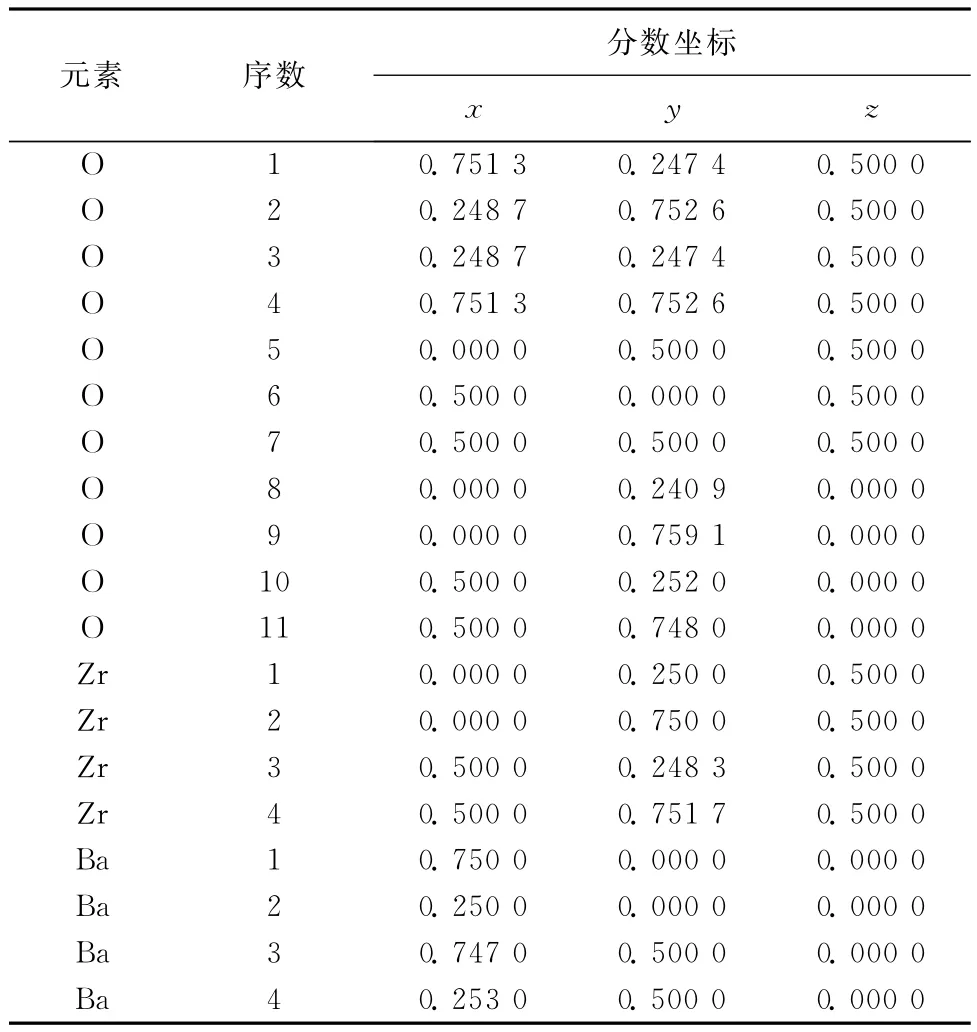
表4 OⅡ空位优化后BaZrO3各原子的坐标Table 4 Atomic coordinates of geometry optimization of OⅡvacancy
表5是理想BaZrO3和OⅡ空位BaZrO3晶体晶格常数和体积的变化.

表5 理想BaZrO3与OⅡ空位BaZrO3晶体晶格常数和体积的变化Table 5 Lattice constants and volume changes of perfect and OⅡvacancy BaZrO3
从表5中可以看出,与理想BaZrO3比较,OⅡ空位BaZrO3晶体的晶格参数均发生明显变化,晶格参数a减少约0.000 2nm,b减少约0.000 4nm,c减少约0.000 2nm,相应的体积由原来的0.298 4nm3减少了3.430 0×10-4nm3.可见,由于OⅡ空位的存在,BaZrO3晶体结构发生了明显变化.
表6为OⅡ空位BaZrO3超胞中各原子轨道上的电子布居数和键布居数.从表6中可以看出,离氧空位较近的Zr原子优化后的电子构型为6s2.425p6.064d2.11,由于氧空位的作用,与理想的 Zr原子相比,其6s态电子数增加0.07,5p轨道增加0.30e,4d轨道增加0.09e;而离氧空位较远的Zr原子与理想的Zr原子差别不大.从键的布居数可知,Zr原子与z轴上相应O原子的键长为0.210nm,重叠部分最大,而与x和y轴周围的O原子重叠部分较小,相互作用比较弱.

表6 OⅡ空位BaZrO3超胞中各原子轨道上电子布居数和键布居数Table 6 Atomic and bond populations on atomic orbits of OⅡvacancy BaZrO3
OⅡ空位BaZrO3晶体中,离氧空位近的Ba原子优化后的电子构型为6s1.865p6.034d0.88,与理想的Ba原子相比,6s轨道电子数减少0.09,5p轨道增加0.02e,而4d轨道增加0.22e;远离氧空位的Ba原子与理想的Ba原子几乎没有差别.在xOy面上,Ba原子与离氧空位远的O原子重叠较少.其他情况的Ba原子与O原子重叠部分很大,说明它们之间的相互作用比较大.
O原子在氧空位中的电子变化不大,2s态几乎不参与成键,而2p轨道的电子分别与Zr原子和Ba原子发生很强的作用.
由于每个O原子与Ba原子和Zr原子之间的相互作用不尽相同,造成BaZrO3晶体的晶格参数发生了变化.
2.2.2 态密度及分态密度分析
图6是OⅡ空位BaZrO3晶体的态密度和分态密度.从图6中可以看出,由于氧空位的影响,晶体中各个原子态密度发生了变化,11个O原子分为6种,Zr原子和Ba均分为2种.各个O原子变化很细微,与布居数反映的相一致.能量低于-10eV的各个态离域性很强,几乎不参与成键.OⅡ空位BaZrO3的价带主要由O原子2p轨道和Zr原子4d轨道电子贡献,而导带主要由Zr原子4d轨道电子构成.
图7是理想BaZrO3与OⅡ空位BaZrO3晶体的态密度图.可以看出,与理想BaZrO3晶体相比,OⅡ空位BaZrO3晶体的态密度峰均向低能方向迁移,且导带和价带区域的杂化相对变弱,说明由于OⅡ空位的存在,晶体中的各原子之间的共价键减弱.
图8是理想BaZrO3与OⅡ空位BaZrO3的能带结构图,从图中可以看出,与完美的BaZrO3晶体比较,OⅡ空位BaZrO3的价带与导带之间的能隙减小为2.592 43eV,导带与价带均向低能级移动,而价带与导带的带宽变窄,这与态密度的变化趋势相一致.
3 结论
本研究采用建立在密度泛函理论基础上的Castep软件包,在广义梯度近似下,利用超软赝势对理想BaZrO3晶体和OⅡ空位BaZrO3晶体的布居数、态密度和能带分布等进行自洽计算.结果显示:在OⅡ空位BaZrO3晶体中,O原子与各个Zr原子和Ba原子之间相互作用所形成的键的键长发生了不同程度的变化,晶体的晶格参数a,b和c各不相同.

图6 OⅡ空位BaZrO3晶体的态密度和分态密度图Figure 6 DOS and PDOS of OⅡvacancy BaZrO3

图7 理想BaZrO3与OⅡ空位BaZrO3晶体的态密度图Figure 7 DOS of perfect and OⅡvacancy BaZrO3

图8 理想BaZrO3与OⅡ空位BaZrO3的能带结构图Figure 8 Band structures of perfect and OⅡvacancy BaZrO3
[1] Wang Y X,Arai M,Sasaki T,et al.First-principles study of the(001)surface of cubic CaTiO3[J].Phys Rev B,73(3):1-6.
[2] 唐春红,蔡孟秋,尹真,等.铁电体SrBi2Nb2O9电子能带结构的第一性原理[J].物理学报,2004,53(9):2931-2935.
[3] Astala R,Bristowe P D.Ab initio study of the oxygen vacancy in SrTiO3[J].Modelling Simul Mater Sci Eng,2001,(9):416-422.
[4] Cai M Q,Zhang Y J,Yin Z,et al.First-principles study of structural and electronic properties of BaTiO3(001)oxygenvacancy surfaces[J].Phys Rev B,2005,72(7):075406-1-075406-6.
[5] Zhang C,Wang C L,Li J C,et al.Surface rumples and band gap reductions of cubic BaZrO3(001)surface studied by means of first-principles calculations[J].Chin Phys Soc,2008,17(1):274-279.
[6] Shimada T,Umeno Y,Kitamura T.Ab initio study of stressinduced domain switching in PbTiO3[J].Phys Rev B,2008,77(9):094105-1-094105-6.
[7] 倪建刚,刘诺,杨果来,等.第一性原理研究BaTiO3(001)表面的电子结构[J].物理学报,2008,57(7):4434-4439.
[8] Eglitis R I,Vanderbilt D.Ab initio calculations of BaTiO3and PbTiO3(001)and(011)surface structures[J].Phys Rev B,2007,76(15):155439-1-155439-9.
[9] Wahl R,Vogtenhuber D,Kresse G.SrTiO3and BaTiO3revisited using the projector augmented wave method:Performance of hybrid and semilocal functionals[J].Phys Rev B.2008,78(10):104116-1-104116-10.
[10] 薛卫东,陈召勇,杨春,等.四方相BaTiO3铁电性的第一性原理研究[J].物理学报,2005,54(2):857-861.
First-principles study on OⅡ vacancy in BaZrO3
SONGZhaotao1,ZHAOHui1,2,CHENTianchi1,DENGXiaobing1
(1.College of Physics and Electronic Information,Tianjin Normal University,Tianjin 300387,China;
2.Department of Physics,Anshan Normal University,Anshan 114005,Liaoning Province,China)
The OⅡvacancy in BaZrO3is studied by first-principles.There are interactions of O atoms with Zr and Ba atoms owing to OⅡvacancy,and atomic relaxation changes the crystal structure,so lattice constants ofa,b,care different.By analyzing the population,density of states and band structure,it is found that OⅡ vacancy would change the crystal structure of BaZrO3.
first-principles;BaZrO3crystal;OⅡvacancy;population;density of states;band structure
O77+1
A
1671-1114(2011)01-0035-07
2009-11-04
辽宁省自然科学基金(20082192)
宋兆涛(1983—),男,硕士研究生.
赵 辉(1965—),男,教授,博士,主要从事凝聚态理论和计算物理方面的研究.
(责任编校 纪翠荣)
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
人工晶体学报(2021年10期)2021-11-26
陶瓷学报(2019年5期)2019-01-12
读者欣赏(2014年6期)2014-07-03
语文知识(2014年2期)2014-02-28

- 天津师范大学学报(自然科学版)的其它文章
- 可微C0-半群的谱
- 绝对破产下的总负持续时间