
锌依赖的组蛋白去乙酰化酶选择性抑制剂研究进展
2010-12-08 06:57:02刘红椿杜晓光耿美玉
中国药理学通报 2010年8期
刘红椿,杜晓光,耿美玉,
近年来,组蛋白去乙酰化酶(HDAC)作为药物靶点吸引了众多的关注,并且已经有HDAC抑制剂Vorinostat和Depsipeptide被美国FDA批准以皮肤T淋巴细胞瘤(CTCL)为适应症而上市应用[1],在实体瘤治疗中的应用也处于临床试验阶段。这标志着HDAC作为新颖药物靶标的概念验证性研究阶段的结束,也预示着HDAC抑制剂作为抗肿瘤药物具有广阔的开发前景。
尽管目前关于HDAC抑制剂的设计研究及作用机制研究越来越多,但是HDAC抑制剂抗肿瘤作用的明确机制尚未阐明。目前人们提出的作用机制很多,包括促凋亡、促分化、细胞周期阻滞、抑制DNA损伤修复、上调肿瘤抑制基因、下调生长因子、氧化应激等等[2-5],但这些作用机制往往依赖于细胞类型、实验条件及特定的化合物。这种多个作用机制现象的出现要归因于表观遗传变化的广泛性,如组蛋白的乙酰化和去乙酰化能够影响多种基因的转录。更重要的是,HDACs不仅仅能够催化组蛋白的去乙酰化,而且能够催化其他一些重要蛋白(如Hsp90、Tubulin等)和转录因子(p53、STAT1等)的乙酰化。现有的HDAC抑制剂如Vorinostat及其他一些处于临床研究的侯选化合物往往是泛抑制剂[6],能同时抑制多个HDAC家族成员。而这些不同亚型的HDAC往往具有不同的定位,在细胞功能调控上发挥着不同的作用,并且与不同的疾病类型相关[7-8]。这种作用机制的广泛性使得 HDAC 泛抑制剂如 Vorinostat、TSA、Belinostat、Panobinostat、LAQ824、JNJ-26481585等会破坏多个依赖于蛋白乙酰化的细胞内过程,不仅影响肿瘤的生长和转移[9],而且会干扰正常的生理功能,使得HDAC泛抑制剂存在潜在的毒副作用。
因此选择性的HDAC抑制剂不仅能够降低毒副作用,而且能为各亚型的生物学功能研究提供探针,具有深远的意义。本文将对HDAC选择性抑制剂进行综述。
1 HDAC家族主要成员的定位和功能
HDAC是一个大的酶家族,其成员目前已知有18个不同的亚型,按种系分为 4 大类:Ⅰ(HDAC 1、2、3、8)、Ⅱ(HDAC 4、5、6、7、9、10)、Ⅲ(SIRT1-SIRT7)和Ⅳ(HDAC 11)[10]。其中Ⅰ、Ⅱ、Ⅳ为经典家族,是 Zn2+-依赖性的HDAC,而Ⅲ属于Sirtuin家族,是NAD+-依赖的HDAC。目前临床研究的绝大多数HDAC抑制剂能够抑制HDAC的多个亚型,这些亚型往往属于Zn2+-依赖性的HDAC家族。而针对NAD+-依赖的HDAC的抑制剂目前研究较少,在此不予涉及。
不同亚型HDAC通常具有不同的基因表达模式、细胞定位和生物学功能。目前研究发现[11],HDAC 1、2、3主要定位在细胞核内,在多种组织细胞中表达,并在多种肿瘤细胞中过表达,且与不良预后有关。HDAC 6主要定位于细胞质,而HDAC 4、5、7和9主要穿梭于细胞核与细胞质之间,只在部分组织细胞中表达,但是有研究表明,HDAC5的表达预示着较好的预后,这提示在肿瘤治疗中最好不要抑制此类HDAC亚型。研究表明,在不同类型的肿瘤中HDAC的表达亚型和表达量不同。这种肿瘤选择性的HDAC表达是亚型选择性HDAC抑制剂应用的病理学基础,也提示HDAC抑制剂的选择性不仅能增强药效,而且能降低毒副作用。这些发现也是促使人们开发选择性HDAC抑制剂的原因之一。见Tab 1。
2HDAC选择性抑制剂
应用RNA干扰技术和动物基因敲除技术研究显示,Ⅰ类和Ⅱa类中的一些HDAC亚型,可能是抗肿瘤作用相关的靶标。其中对HDAC 1、2、3的抑制会引起细胞周期阻滞,产生细胞增殖抑制作用[15];对HDAC 4的抑制会影响DNA损伤修复的过程[16];对HDAC 6的抑制会影响Tubulin的乙酰化,降低肿瘤细胞的转移能力[17]等。随着HDAC各个亚型的具体调控机制、生物学功能以及与肿瘤类型关系的深入研究,有针对地选择某些HDAC亚型的选择性抑制剂对个体化治疗具有重要意义,也将更受人们的青睐。
2.1 HDAC1选择性抑制剂 HDAC 1是1996年由美国哈佛大学Taunton等[18]发现的第一个哺乳动物的组蛋白去乙酰化酶,它是多种蛋白质复合物的催化亚单位,参与组蛋白和非组蛋白的去乙酰化。HDAC 1的表达在肺癌、乳腺癌、前列腺癌和胃肠道癌等都有明显升高,被认为是肺癌、乳腺癌的预后指标[10]。研究发现[19],HDAC 1 能调控周期相关蛋白的转录,HDAC 1的抑制主要通过上调p21的表达导致细胞周期的阻滞。
MS-275是1999年由 Suzuki等[20]合成的苯甲酰胺类HDAC 1抑制剂。与泛抑制剂的作用机制不同,MS-275不是以Zn2+为结合靶标,而是以活性口袋最狭窄的部位Phe141和Phe198相对的2个苯环为靶点,阻塞了HDAC的生理底物(组蛋白N端Lys乙酰化侧链)伸向催化中心的通道,因此其对亚型选择性明显好于Zn2+为结合靶点的抑制剂。MS-275通过增加肿瘤细胞中周期阻滞蛋白p21的水平,抑制细胞增殖,促进细胞分化,在动物实验中口服抗肿瘤活性好而且毒副作用低[21]。目前该药物正在美国进行白血病及实体瘤的临床Ⅱ期研究。另外,临床研究表明,MS-275与DNA甲基转移酶抑制剂5-阿扎胞苷联用,对第一次化疗失败的高分化的非小细胞肺癌患者有治疗作用,显示了良好的抗肿瘤作用。
2.2 HDAC1/2选择性抑制剂 HDAC 2与HDAC 1具有高达82%的同源性,且二者的作用机制相似,单独酶不具有HDAC活性,必须与辅因子形成复合物才能有去乙酰化酶活性。HDAC 1和HDAC 2可直接与DNA结合蛋白如YY1、SP1等结合。HDAC 2在抑制凋亡中具有重要作用[19]。
2.2.1 Depsipeptide Depsipeptide,又名 FK228、FR901228、NSC630176,是一来源于天然产物的环肽类化合物,其对HDAC 1和HDAC 2的选择性相似。计算机模拟的结果表明,Depsipeptide的硫醇基团可以通过一个水分子与Zn2+结合[22]。Depsipeptide对多种血液肿瘤和实体瘤都具有明显的杀伤作用。目前Depsipeptide已经获得FDA批准用于皮肤T细胞淋巴瘤的治疗,其针对慢性淋巴细胞白血病、急性髓样白血病和其他实体瘤的研究正处于临床阶段[23]。
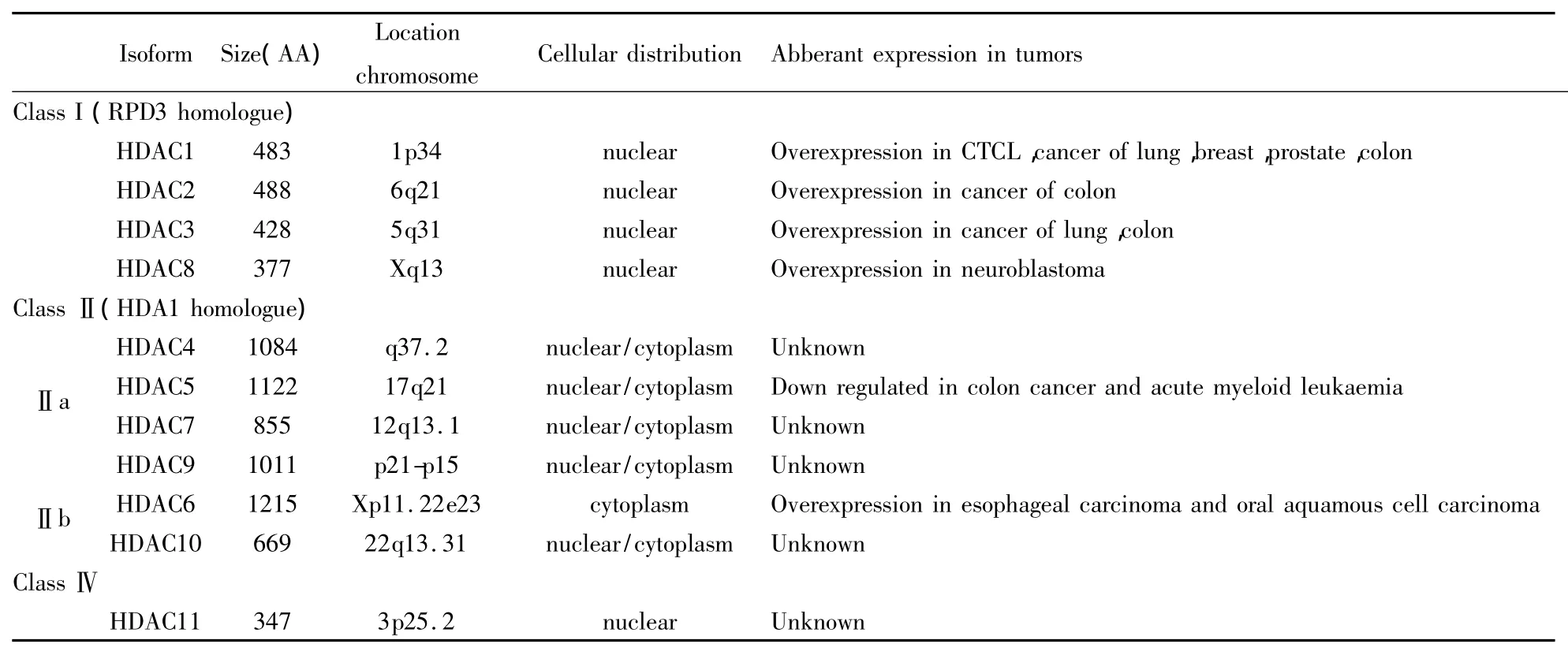
Tab 1 Properties of zinc dependant HDACs
2.2.2 MGCD103 MGCD0103是一种新型的苯氨基苯甲酰胺类HDAC抑制剂[24]。MGCD0103可以抑制多种肿瘤细胞的生长,比泛抑制剂SAHA和HDAC 1特异性抑制剂MS-275的增殖抑制活性更强[24]。而另有研究表明[25],MGCD0103主要抑制HDAC 1的活性,同时对HDAC 2、3、11都有一定的抑制作用,故其选择性还有待进一步的验证。MGCD0103口服有效,且显示了良好的药效和药代动力学特性,目前正处于多项Ⅱ期临床研究,是治疗血液学疾病和淋巴组织增生疾病的一个比较有前景的化合物[26]。
2.3 HDAC 8选择性抑制剂 Buggy等[27]在2000年发现了HDAC的第8个成员——HDAC 8,其在心脏、肺、肾脏和胰腺中均有表达,在肝中的表达量最高。HDAC 8与HDAC 3的同源性最高,达到34%。Knockdown实验表明HDAC 8对细胞增殖是必须的[28]。HDAC 8在神经细胞瘤中有异常表达,选择性地抑制其活性,可诱导神经细胞瘤的分化[13],提示应用选择性HDAC 8抑制剂可治疗神经细胞瘤。
PCI-34051是一个低分子量的异羟肟酸类化合物,能特异性抑制HDAC 8的活性。通过激活PLCγ,PCI-34051能够诱导T淋巴瘤细胞和白血病细胞发生caspase依赖性的凋亡,但是对其他造血细胞和实体瘤细胞无明显影响[29],提示PCI-34051可能发展成为一个高效低毒选择性强的T细胞恶性肿瘤的治疗药物。
2.4 HDAC6选择性抑制剂 HDAC6属于ClassⅡb,其独特之处是拥有两个催化区域:一个起始于第215位氨基酸,另一个起始于第610位,两个催化区高度同源,而且均含有一个与泛素结合的锌指结构区。正常生理条件下,HDAC 6在心、肝、肾、胰等脏器中高表达[30],而在食道癌[31]和口腔鳞状上皮细胞癌中过表达。选择性抑制HDAC 6对细胞的存活影响不大,但是可以明显增加tubulin和HSP90的乙酰化水平[32-33],从而降低细胞的运动能力和加速HSP90客户蛋白的降解。代谢活跃的细胞尤其是肿瘤细胞中产生了大量错误折叠蛋白,HDAC 6能促进错误折叠蛋白降解,故HDAC 6的选择性抑制可以严重地破坏细胞处理错误折叠蛋白的能力,导致细胞内的蛋白“垃圾”迅速积累,进而诱发肿瘤细胞凋亡,而对正常细胞没有明显影响,因此近期HDAC 6的选择性抑制剂备受关注。
2.4.1 Tubacin Tubacin是 Schreiber领导的研究组通过对7392种小分子的筛选得到的HDAC 6的抑制剂,其抑制了哺乳动物细胞的tubulin去乙酰化,但是对组蛋白的乙酰化水平、基因转录和细胞周期进程没有影响,tubacin能增加tubulin的乙酰化,虽然不影响tubulin的稳定性,但是能够降低细胞的运动性[17]。另外tubacin并不抑制Hsp90的去乙酰化,提示tubacin可能特异性抑制HDAC6-SIRT2复合物的形成,而该复合物负责tubulin的去乙酰化[34]。
2.4.2 5-Aryl-2-(trifluoroacetyl)thiophene 该化合物是由Ontoria等[35]研究的HDAC抑制剂,对HDAC 4和HDAC 6的选择性较好,其抑制HDAC 4和HDAC 6的IC50值分别为310 nmol·L-1和 70 nmol·L-1,对 HDAC 1 的 IC50比 HDAC 6 强40多倍,而且对实体瘤细胞抑制效果较好。
2.4.3 含吡啶丙氨酸的羟肟酸类化合物 Schafer等[36]研究发现在羟肟酸上进行吡啶丙氨酸修饰,可以得到选择性的HDAC 6抑制剂5a,其对HDAC 1的IC50是HDAC 6的25倍左右,在细胞水平能够升高tubulin的乙酰化水平,其对肿瘤细胞的细胞毒活性较HDAC泛抑制剂弱,与蛋白酶体抑制剂硼替佐米有协同致死作用,提示联合用药可作为其应用的一种策略。
上述的亚型选择性HDAC抑制剂,从严格意义上讲,并不具有绝对的选择性。一方面是由于HDAC各亚型在很大程度上的同源性,既照顾到抑制活性又兼顾选择性难度较大;另一方面现有的抑制剂在分子水平的酶活评价时,仅有少数能在11个HDAC亚型上进行全面评价,大多数化合物仅选用若干HDAC酶进行筛选,所得数据不能完全确定其亚型选择性,因此全面筛选是亚型选择性抑制剂开发和评测的重要指标。
3 展望
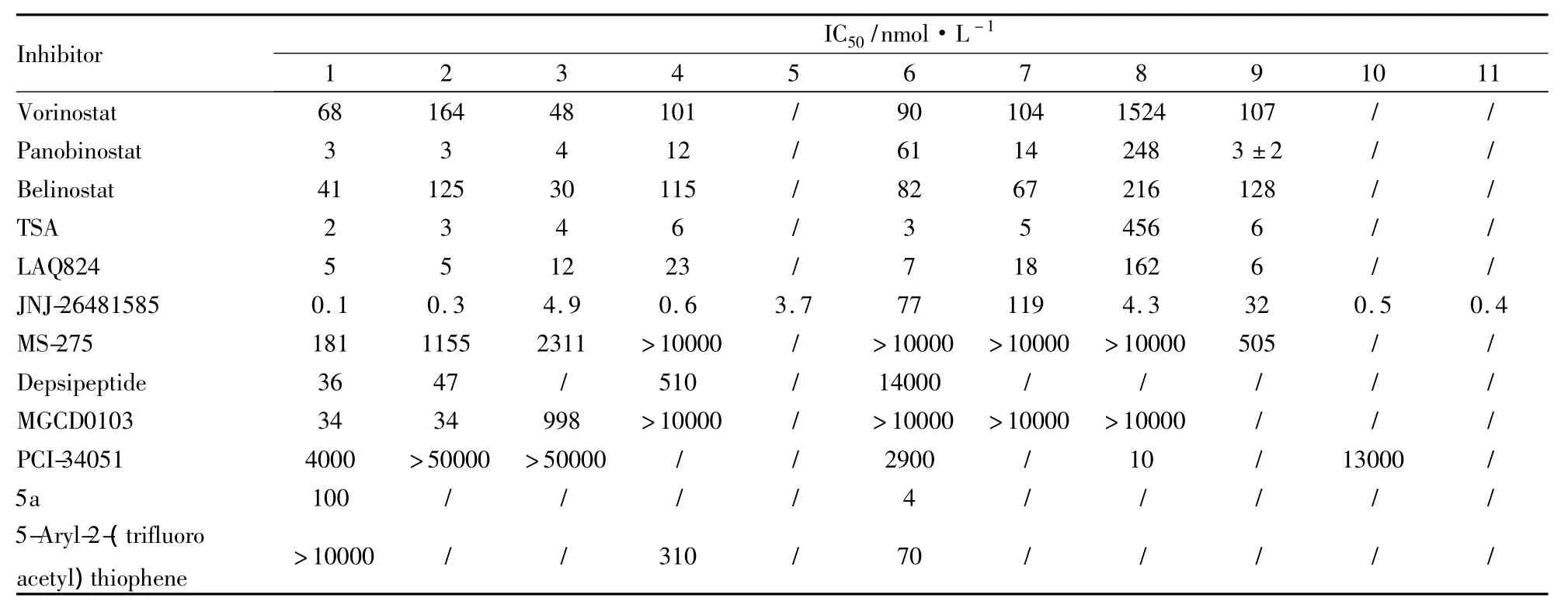
Tab 2 Inhibitors activity toward HDAC enzymes
目前各种HDAC亚型的特异生物学功能、选择性表达的肿瘤类型等都没有完全阐释清楚,但是结合已有的研究成果可以预测亚型选择性的HDAC抑制剂拥有明显的优势,如对肿瘤组织选择性高、毒副作用较低、可与靶向药物的联合应用[37]等,因此亚型选择性的HDAC抑制剂是各大研究机构和药厂开发的目标。尽管目前有很多HDAC的抑制剂处于不同的研发阶段,但是受限于HADC亚型的同源性较高,且大多数亚型没有晶体结构,选择性的HADC抑制剂数量还是相当有限的,而且亚型绝对选择性的抑制剂更是罕见。随着HDAC亚型生物学研究的深入、晶体结构的健全和完善以及计算机模拟的合理化设计,更多的亚型选择性的HDAC抑制剂将会涌现出来。
[1] Shabason J E,Tofilon P J,Camphausen K.HDAC inhibitors in cancer care[J].Oncol(Williston Park),2010,24(2):180 -5.
[2] Rosato R R,Grant S.Histone deacetylase inhibitors:insights into mechanisms of lethality[J].Expert Opin Ther Targets,2005,9(4):809-24.
[3] Xu W S,Parmigiani R B,Marks P A.Histone deacetylase inhibitors:molecular mechanisms of action[J].Oncogene,2007,26(37):5541-52.
[4] Bolden J E,Peart M J,Johnstone R W.Anticancer activities of histone deacetylase inhibitors[J].Nat Rev Drug Discov,2006,5(9):769-84.
[5] Glaser K B.HDAC inhibitors:clinical update and mechanismbased potential[J].Biochem Pharmacol,2007,74(5):659 -71.
[6] 李新刚,吴 青.TSA抑制NB4细胞去乙酰化酶活性并促进细胞周期素激酶抑制剂表达[J].中国药理学通报,2005,21(2):165-8.
[6] Li X G,Wu Q.The inhibition of histone deacetylase and expression promotion of cyclin dependent kinase inhibitor in raji cells by trichostatin A[J].Chin Pharmacol Bull,2005,21(2):165 -8.
[7] 唐丽琴,魏 伟.组蛋白去乙酰化酶抑制剂与炎症免疫性疾病[J].中国药理学通报,2007,24(1):1 -4.
[7] Tang L Q,Wei W.Histone deacetylase inhibitors and inflammatoryimmune diseases[J].Chin Pharmacol Bull,2007,24(1):1 -4.
[8] 孟 玫,姜军梅,尹晓燕,等.苯丁酸钠对两种人肝癌细胞抑癌基因表达的影响[J].中国药理学通报,2005,21(11):1406-7.
[8] Meng M,Jiang J M,Yin X Y,et al.Effects of Sodium phenylbutyrate on differentiation and induction of antigene in human liver carcinoma cell lines[J].Chin Pharmacol Bull,2005,21(11):1406-7.
[9] Yasui W,Oue N,Ono S,et al.Histone acetylation and gastrointestinal carcinogenesis[J].Ann N Y Acad Sci,2003,983:220 -31.
[10]Bertrand P.Inside HDAC with HDAC inhibitors[J].Eur J Med Chem,2010,45(6):2095 -116.
[11]Weichert W,Roske A,Gekeler V,et al.Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer:a retrospective analysis[J].Lancet Oncol,2008,9(2):139-48.
[12]Bolden J E,Peart M J,Johnstone R W.Anticancer activities of histone deacetylase inhibitors[J].Nat Rev Drug Discov,2006,5(9):769-84.
[13]Oehme I,Deubzer H E,Lodrini M,et al.Targeting of HDAC8 and investigational inhibitors in neuroblastoma[J].Expert Opin Investig Drugs,2009,18(11):1605 -17.
[14]Sakuma T,Uzawa K,Onda T,et al.Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma[J].Int J Oncol,2006,29(1):117 -24.
[15]Inoue S,Mai A,Dyer M J,et al.Inhibition of histone deacetylase class I but not classⅡis critical for the sensitization of leukemic cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis[J].Cancer Res,2006,66(13):6785 -92.
[16]Kao G D,McKenna W G,Guenther M G,et al.Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response[J].J Cell Biol,2003,160(7):1017 -27.
[17] Haggarty S J,Koeller K M,Wong J C,et al.Domain-selective small-molecule inhibitor of histone deacetylase 6(HDAC6)-mediated tubulin deacetylation[J].Proc Natl Acad Sci USA,2003,100(8):4389-94.
[18] Taunton J,Hassig C A,Schreiber S L.A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p[J].Science,1996,272(5260):408 -11.
[19]Zhu P,Huber E,Kiefer F,et al.Specific and redundant functions of histone deacetylases in regulation of cell cycle and apoptosis[J].Cell Cycle,2004,3(10):1240 -2.
[20]Suzuki T,Ando T,Tsuchiya K,et al.Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives[J].J Med Chem,1999,42(15):3001 -3.
[21]Saito A,Yamashita T,Mariko Y,et al.A synthetic inhibitor of histone deacetylase,MS-27-275,with marked in vivo antitumor activity against human tumors[J].Proc Natl Acad Sci USA,1999,96(8):4592-7.
[22]Furumai R,Matsuyama A,Kobashi N,et al.FK228(depsipeptide)as a natural prodrug that inhibits class I histone deacetylases[J].Cancer Res,2002,62(17):4916 -21.
[23] Shah M H,Binkley P,Chan K,et al.Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors[J].Clin Cancer Res,2006,12(13):3997 -4003.
[24]Khan N,Jeffers M,Kumar S,et al.Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors[J].Biochem J,2008,409(2):581 -9.
[25]Fournel M,Bonfils C,Hou Y,et al.MGCD0103,a novel isotype-selective histone deacetylase inhibitor,has broad spectrum antitumor activity in vitro and in vivo[J].Mol Cancer Ther,2008,7(4):759-68.
[26]Le T C,Siu L L.Promising antitumor activity with MGCD0103,a novel isotype-selective histone deacetylase inhibitor[J].Expert Opin Investig Drugs,2008,17(8):1247 -54.
[27]Buggy J J,Sideris M L,Mak P,et al.Cloning and characterization of a novel human histone deacetylase,HDAC8[J].Biochem J,2000,350 Pt(1):199 -205.
[28]Vannini A,Volpari C,Filocamo G,et al.Crystal structure of a eukaryotic zinc-dependent histone deacetylase,human HDAC8,complexed with a hydroxamic acid inhibitor[J].Proc Natl Acad Sci USA,2004,101(42):15064 -9.
[29] Balasubramanian S,Ramos J,Luo W,et al.A novel histone deacetylase 8(HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas[J].Leukemia,2008,22(5):1026 -34.
[30]李 斌,王济明.组蛋白去乙酰化酶6(HDAC6)与肿瘤[J].临床和实验医学杂志,2006,5(12):2030 -1.
[30]Li B,Wang J M.Histone deacetylase 6 and tumor[J].J Clin Exp Med,2006,5(12):2030 -1.
[31]杨 晋,赵绍林,杨新玲,等.HDAC6在食道癌中表达的初步研究[J].中国实验诊断学,2009,13(11):1577 -1579.
[31]Yang J,Zhao S L,Yang X L,et al.Study on the expression of HDAC6 in esophageal carcinoma[J].Chin J Lab Diagn,2009,13(11):1577-9.
[32]Zhang Y,Li N,Caron C,et al.HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo[J].EMBO J,2003,22(5):1168-79.
[33]Bali P,Pranpat M,Bradner J,et al.Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90:a novel basis for antileukemia activity of histone deacetylase inhibitors[J].J Biol Chem,2005,280(29):26729 -34.
[34]North B J,Marshall B L,Borra M T,et al.The human Sir2 ortholog,SIRT2,is an NAD+-dependent tubulin deacetylase[J].Mol Cell,2003,11(2):437 -44.
[35]Ontoria J M,Altamura S,Di M A,et al.Identification of novel,selective,and stable inhibitors of class II histone deacetylases.Validation studies of the inhibition of the enzymatic activity of HDAC4 by small molecules as a novel approach for cancer therapy[J].J Med Chem,2009,52(21):6782 -9.
[36]Schafer S,Saunders L,Schlimme S,et al.Pyridylalanine-containing hydroxamic acids as selective HDAC6 inhibitors[J].Chem Med Chem,2009,4(2):283 -90.
[37]Frew A J,Johnstone R W,Bolden J E.Enhancing the apoptotic and therapeutic effects of HDAC inhibitors[J].Cancer Lett,2009,280(2):125-33.
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08 00:39:50
第一财经(2019年8期)2019-08-26 17:53:46
中国组织化学与细胞化学杂志(2016年4期)2016-02-27 11:15:53
中国病理生理杂志(2015年8期)2015-12-21 12:38:10
哈尔滨医药(2015年2期)2015-12-01 03:57:13
学习月刊(2015年14期)2015-07-09 03:37:48
中国当代医药(2015年30期)2015-03-01 02:08:19
中国当代医药(2015年16期)2015-03-01 02:03:13
物理化学学报(2015年5期)2015-02-28 17:34:47
癌变·畸变·突变(2014年2期)2014-03-01 04:39:41
