
62例视神经脊髓炎的临床特点分析
2010-11-26 10:12李晓晖魏世辉
中国中医眼科杂志 2010年2期
李晓晖 魏世辉
视神经脊髓炎(NMO)是一种免疫介导、主要累及视神经和脊髓的中枢神经系统脱髓鞘疾病。该病于1894年首先由Devic和Gault总结报道,又称Devic病或Devic综合征〔1〕。NMO临床上主要表现为双眼同时发生或先后发生的视神经炎和脊髓炎。我们回顾研究了我院眼科及神经内科2000年1月~2009年7月间收治的62例视神经脊髓炎病例的临床资料,着重对其眼部表现及相关检查结果进行分析。
1 对象和方法
1.1 对象
62例视神经脊髓炎病例均依据Wingerchuck〔2〕等在1999年提出的新的NMO诊断标准确定,其具体内容为:必要诊断标准(1)视神经炎;(2)急性脊髓炎;(3)无视神经及脊髓以外的受累。主要支持条件:(1)发病时颅脑MRⅠ阴性(正常或不符合MS影像学的诊断标准);(2)脊髓MRⅠ≥3个椎体的T2信号;(3)CSF 细胞数增多(WBC>50mm3)或中性粒细胞>5mm3。 次要支持条件:(1)双侧视神经炎;(2)至少一眼视力持续低于20/200;(3)和疾病相关的一个或一个以上肢体持续无力(MRC2级或以下)。上述条件中符合全部必须诊断标准和1个主要支持条件或2个次要支持条件,并除外其他自身免疫疾病所致的视神经脊髓损伤可能时,可考虑NMO。
1.2 方法
首诊患者常规眼科检查及全身体检均由主治级别以上医师完成,并查阅和详细记录外院相关检查内容,对能合作者完善相关各项检查,包括标准对数视力表、气动眼压仪、101视野仪30°视野检查、视觉诱发电位及MRⅠ等检查,常规腰穿检查CSF。
2 结果
本组 62例患者中,男 15例(24.2%),女 47例(75.8%)。 发病年龄 3~69 岁,平均(39.44±4.28)岁,20~59岁占发病人数74%。首次发病27例(43.55%),复发病例35例(56.45%)。62例NMO均表现为急性或亚急性起病的单侧或双侧视神经或脊髓损害症状而无其他部位受损。病程最短30天,最长20年。
2.1 视神经损害
视神经受累症状为先者35例(56%),脊髓受累为首发症状者为23例(38%),视神经与脊髓症状同时出现者4例(6.45%)。视神经病变症状与脊髓病变症状间隔最短2天,最长18年,平均(1.94±0.95)年。双侧视神经症状发病者47例(75.8%),其中双眼贯序发病者37例(78.7%),相隔最短5天,最长6年,平均(1.46±0.79)年。视神经首先受累的35例病例中,单眼受累14例(23%),双眼受累48例(77%),均表现为视力不同程度下降。双眼受累病例中,患眼视力下降0.8~无光感,其中患眼无光感11例(23%),眼前手动22例 (45%),其余患眼视力为0.01~0.8。患眼均表现为不同程度视野缺损。受累眼均表现为瞳孔散大,RAPD (+)。其中26例首诊眼科,仅出现单侧视神经受累,未出现脊髓症状前12例(35.3%)诊断为球后视神经炎,9例(26.5%)诊断为视神经乳头炎,3例(8.8%)诊断为视神经萎缩,2例正确诊断为NMO,漏诊率达93%。
2.2 脊髓损害
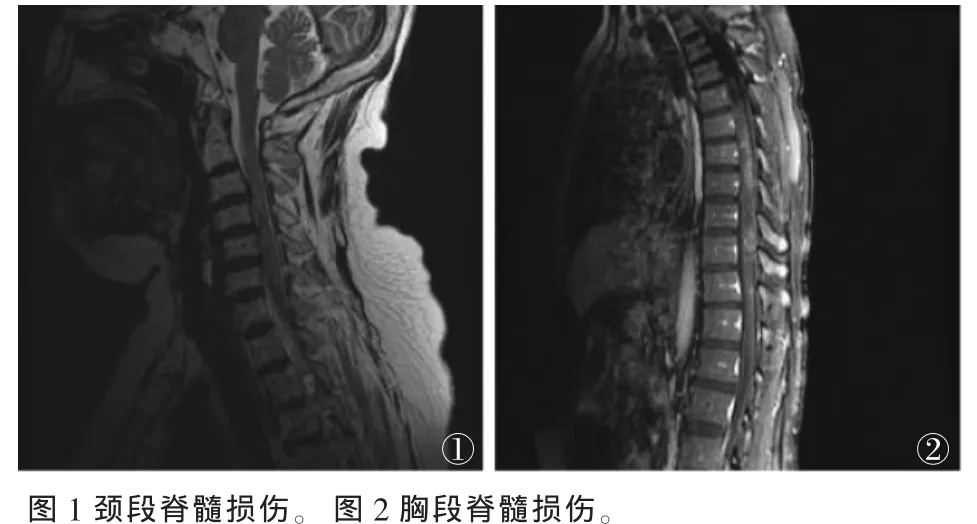
62例NMO病例均有不同程度脊髓损害,其中表现为双下肢异常者27例(43.55%),双上肢异常者 3例(4.84%),一侧肢体异常者 15例(24.19%),四肢异常者12例(19.35%),面部或手指麻木症状者6例(9.68%),伴尿便障碍者19例(30.65%)。脊髓损伤平面在颈段(图 1)者35例(56.45%),胸段(图 2)者 17例(27.42%),腰段者 3 例(4.84%),未见异常7例(11.29%)。在脊髓MRⅠ图像中,可见脊髓内增粗、水肿等不均匀密度影弥漫分布的斑片状或点状长T1、T2信号,病灶常累及3个以上脊髓节段。
2.3 CSF常规及生化检查
62例NMO病例中36例通过腰穿行CSF检查,其中6例行CSF检查3次。所有病例CSF外观未见异常,均呈无色、透明状,脑脊液压力均在60~200mmH2O之间,全部CSF中糖和氯化物含量在正常范围。21例CSF白细胞数异常(正常参考值为0~5个/mm3),17例蛋白含量增高(参考值为0.15~0.45g/L)。
3 讨论
NMO眼部临床表现为双眼同时或反复发生的视神经炎,病程进展迅速,可数日数小时后发展为完全失明。本组中32例(51.61%)(44只眼)矫正视力≤0.01,属重症 NMO,此结果与戚晓昆〔3〕报道的 NMO的视神经炎首次发作达高峰时,约40%的患眼几近失明相符。因脊髓炎一般迟于视神经炎出现,使得大部分患者因视力显著下降而首诊眼科,NMO早期病变如仅表现为视神经炎,与单纯视神经炎很难鉴别。本组35例首发眼部病例中24例被诊断为球后视神经炎、视神经乳头炎及视神经萎缩。在首次视神经炎发作后5年和10年患者转变为临床确诊的NMO的平均几率是30%和38%,其中伴有异常MRⅠ信号的视神经炎在5年和10年转化为NMO的危险性分别为51%和56%〔4〕。因此,眼科医生应该告知患者,视神经炎可能会继发NMO,并建议他们去神经科进行早期诊断治疗〔5〕。 目前,随着 Lennon〔6〕等在血清中发现的NMO特异性自身抗体NMO-ⅠgG及NMO特异性抗体水通道蛋白4抗体 (APQ4)的发现,相关检测对NMO诊断的特异性和敏感性都有很大的提高,且其抗体滴度水平有助于预测疗效及预后。我院眼科近期已开展了对NMO及ⅠDON病人血清进行APQ4检测,相关研究正在进行中。
NMO的主要病理改变为脱髓鞘、硬化斑和坏死,伴血管周围炎性细胞浸润〔7〕。视觉诱发电位主要反映神经纤维脱髓鞘改变,对病情程度的判断有一定价值。本组62例病人中行视觉诱发电位检查43人,异常结果为42人,表现为P100潜伏期延长,波幅降低。随着病情加重,波形消失。说明VEP从电生理方面对神经功能进行分析,轻微神经病变即有显著发现。本组中有3例首发脊髓病人在未出现视力下降早期即已表现为VEP异常,从而确立了NMO诊断,对其早期诊断价值,与王江桥〔8〕文献报道一致。本组62人中,有55人有视神经颜色记录,病程超过1个半月以上患者出现视神经萎缩者达29人,占53%,这与魏世辉〔9〕报道的视神经炎发生后 4~6周才会发生视神经萎缩相一致。
4 小结
归纳视神经脊髓炎的临床特征:视神经脊髓炎的发病以女性较为多见,常在40岁左右发病,早期多表现为视神经症状 (球后视神经炎和视乳头炎),脊髓MRⅠ显示,脱髓鞘多发生于颈段和胸段(病灶大于3个椎体节段),视觉诱发电位检查和血清水通道蛋白4检测是确诊敏感指标,视神经萎缩是常见后遗症。
1 Jaoob,Matiello M,Wingerchuk DM,et al.Neuromyelitis optica:changing concepts[J].J Neuroimmunol,2007,187(1-2):126-138.
2 Wingerchuk DM,Hogancamp WF,O′Brien PC,et al.The clinical course of neuromyelitis optica (Devic's syndrome) [J].Neurology,1999,53(5):1107-1114.
3 戚晓昆.视神经脊髓炎的诊断与治疗[J].中国全科医学,2007,10(12):955-956.
4 Optic Neuritis Study Group.High and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis[J].Arch Ophthalmol,2003,121:944-949.
5 魏世辉,周必业.关于神经眼科疾病诊断中的几个热点问题[J].眼科,2006,15(6):366-368.
6 Lennon VA,Kryzer TJ,Pittock SJ,et al.ⅠgG marker of opticspinal mutiple sclerosis binds to the aquaporin-4 water channel[J].J ExpMed,2005,202(4):473-477.
7 Wingerchuk DM.Neuromyelitis optica[J].Ⅰnt MS J,2006,13(2):42-50.
8 王江桥.视神经脊髓炎的临床与VEP和MRⅠ表现 [J].眼科研究,2003,21(5):533-534.
9 魏世辉.进一步提高对视神经炎与多发性硬化的认识 [J].眼科,2007,16(6):361-362.
猜你喜欢
中国现代药物应用(2022年17期)2022-10-17
当代医药论丛(2021年24期)2022-01-20
昆明医科大学学报(2021年4期)2021-07-23
中国医科大学学报(2021年4期)2021-05-18
中国全科医学(2019年18期)2019-07-03
中国中医急症(2019年10期)2019-05-21
中国医疗器械信息(2018年21期)2018-12-10
中国内镜杂志(2018年8期)2018-08-31
中国医药指南(2017年15期)2017-01-16
中国卫生标准管理(2015年17期)2015-01-26
