
急性血糖波动对心肌细胞核因子κB及其抑制因子IκB表达的影响△
2010-11-22 06:41中国医科大学盛京医院内分泌科沈阳110004
陕西医学杂志 2010年3期
中国医科大学盛京医院内分泌科(沈阳 110004)
赵 晟 张 微 李 艳 韩 萍*
糖尿病心肌病变(DM)是由糖尿病高血糖状态所导致的一种心脏疾病[1]。氧化应激是所有糖尿病并发症损害发生的主要因素[2],有证据显示急性的血糖波动可以导致机体内氧化应激的产生[3]。本研究采用给Wistar雄性大鼠间断或持续地静脉输注50%葡萄糖溶液建立急性血糖波动或持续性动物模型,探讨血糖波动诱导的氧化应激反应对心肌细胞的影响。
材料和方法
1 实验材料
1.1 实验动物:选取7周健康雄性Wistar大鼠18只,体重250~280g,由中国医科大学实验动物中心提供,清洁度Ⅰ级,随机被分为3组,分别为对照组、血糖波动组、持续高血糖组,常规饲料自由饮水,温度 22±1℃,湿度 50%左右,明 /暗周期为12h。
1.2 主要试剂:兔抗大鼠NF-κB多克隆抗体、兔抗大鼠 IκB多克隆抗体免疫组化 SABC染色试剂盒,均购自武汉博士德公司。血清MAD、SOD检测试剂盒,购自南京建成生物工程研究所。
2 实验方法
2.1 建立血糖波动动物模型:大鼠适应实验室环境 3~5d后,将导管分别植入右颈内静脉用于输液和左颈动脉用于采血。术后3d,进行输液。分别予对照组 0.9%生理盐水,使血糖维持在 5mmol/L左右;予血糖波动组 50%葡萄糖注射液间断输注,使血糖在5mmol/L与20mmol/L之间波动,每 2h变动血糖值,持续2h,见表1。予持续高血糖组50%葡萄糖注射液持续输注,使血糖维持在20mmol/L左右。每小时检测血糖,所有实验大鼠输液时间在48h。
2.2 标本采集:输注相应液体 48h后,取血 5ml,3000转 /分离心 15min,取上清于-70℃保存备用。摘取左心室心肌,置于4%含 0.1%DEPC的多聚甲醛中固定。
2.3 指标检测:①血清指标:用比色法法测定血清MDA、SOD;血糖测定采用葡萄糖氧化酶法测定血糖(BIOSEN5030,德国)。②观察心肌细胞内 NF-κB及IκB表达:用免疫组化的方法测定心肌细胞内 NF-κB、IκB。
2.4 结果判定:利用 Axioplan 2 imaging显微图像分析系统测定 NF-κB、IκB阳性着色面积(%)和平均吸光度,以阳性着色面积×平均吸光度计算NF-κB在细胞核中的相对表达量,IκB在细胞质中的相对表达量。
结 果
1 对照组、血糖波动组、持续性高血糖组血糖测定值,见表1,表2。
表1 血糖波动组高峰平台期血糖值与对照组、持续性高血糖组同时相血糖值的比较(±s)

表1 血糖波动组高峰平台期血糖值与对照组、持续性高血糖组同时相血糖值的比较(±s)
持续高血糖组 20.1±14 21.5±4.1 21.3±0.2 19.6±1.6 20.1±1.0 21.7±2.3 19.9±0.3 19.4±1.6 20.7±3.2 19.7±1.6 21.9±2.0 20.7±3.0
表2 血糖波动组低谷平台期血糖值与对照组、持续性高血糖组同时相血糖值的比较(±s)
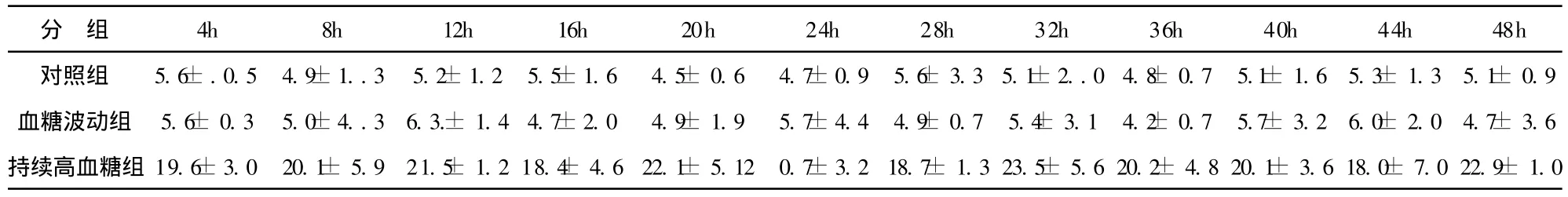
表2 血糖波动组低谷平台期血糖值与对照组、持续性高血糖组同时相血糖值的比较(±s)
血糖波动组 5.6±0.3 5.0±4..3 6.3.±1.4 4.7±2.0 4.9±1.9 5.7±4.4 4.9±0.7 5.4±3.1 4.2±0.7 5.7±3.2 6.0±2.0 4.7±3.6持续高血糖组 19.6±3.0 20.1±5.9 21.5±1.2 18.4±4.6 22.1±5.12 0.7±3.2 18.7±1.3 23.5±5.6 20.2±4.8 20.1±3.6 18.0±7.0 22.9±1.0
2 血清 MDA、SOD表达水平,见表3。血糖波动组及持续性高血糖组血清中 MDA、SOD表达水平明显高于对照组(P<0.05),而且血糖波动组血清中 MDA、MDA/SOD比值均高于持续性高血糖组(P<0.01);血糖波动组MDA/SOD比值显著高于对照组及持续性高血糖组(P<0.01),持续性高血糖组 MDA/SOD比值高于对照组但是无统计学差异(P>0.05)。

表3 对照组、血糖波动组及持续性高血糖组血清 MDA(nmol/ml)、SOD(U/ml)水平
血糖波动组、持续性高血糖组心肌细胞核内 NF-κB、IκB的表达明显高于对照组(P<0.05),血糖波动组与持续性高血糖组无统计学差异(P>0.05);血糖波动组、持续性高血糖组心肌细胞的NF-κB/IκB明显高于对照组(P<0.05),血糖波动组心肌细胞内表达的NF-κB/IκB比值低于持续性高血糖组,但是无统计学差异(P>0.05)。
3 心肌细胞内 NF-κB、IκB及NF-κB/IκB比值表达,见表4。

表4 对照组、血糖波动组及持续性高血糖组血清MDA、SOD水平
讨 论
NF-κB是一种具有多向性的转录因子,能被多种物质刺激激活。NF-κB蛋白 N-末端具有 Rel蛋白同源结构域(Rel homology domain,RHD),该结构域是NF-κB二聚化的活性位点、与定位细胞核 DNA结合位点和IκB相互作用位点,蛋白 C末端拥有 IκB特征性结构域-锚蛋白重复序列[3,4]。当细胞未受到外界刺激时,IκB与NF-κB结合形成复合体,存在于细胞质内;当细胞受到外界刺激时如细胞因子等,细胞质中的IκB激酶(IκB kinase,IKK)发生积聚并被激活,活化的IKK能使 IκB磷酸化,之后 IκB与NF-κB分离,并被降解,同时NF-κB被激活,而进入细胞核促进炎症因子如细胞间粘附因子(Intercellular adhesion molecular-1,ICAM-1)等基因转录表达。
本研究对 Wistar大鼠心肌细胞的研究过程中,发现血糖波动组及持续性高血糖组的心肌细胞核NF-κB表达明显高于对照组(P<0.05)。此外,发现血糖波动组及持续性高血糖组大鼠血清中MDA、MDA/SOD的水平显著高于对照组(P<0.05),而血糖波动组表达又显著高于持续性高血糖组(P<0.01),反映血糖波动和持续性高血糖都能诱导机体出现强烈的氧化应激反应,而且血糖波动诱导机体氧化应激的作用更强。血糖波动和持续性高血糖通过诱导实验雄性Wistar大鼠体内氧化应激反应增强,激活心肌细胞内NF-κB,从而入核表达。Osorio-Fuentealba C等[5,6]在缺氧的心肌细胞中发现活性氧簇(Reactive oxidative species,ROS)明显升高,ROS激活了NF-κB,启动介导的细胞信号转导通路。
NF-κB作为典型的多向性转录因子,在许多的免疫应答反映的信号途径中发挥关键性作用。NF-κB能被多种刺激如细胞因子等激活,活化的NF-κB介导多种促炎症的细胞因子和粘附分子的表达和合成[7]。心肌细胞内 NF-κB细胞信号转导通路的功能非常复杂,除促进促炎症的细胞因子的表达外,NF-κB还能激活其他多种基因的表达,继而影响心肌细胞的生长、收缩、死亡及细胞外基质重塑等病理生理过程[8]。
近年来研究显示,NF-κB活化导致细胞内诱导型一氧化氮合酶(Inducible nitric-oxide synthase,iNOS)表达,促进细胞内氧化应激反应增加作用[9]。而大量ROS的蓄积可激活 JNK介导的线粒体依赖性细胞凋亡信号途径,从而诱导细胞发生凋亡[10~12]。糖尿病心肌细胞存在明显的凋亡现象,而影响凋亡的原因之一就是高糖。本研究发现血糖波动能导致机体 ROS增多及心肌细胞内 NF-κB激活,后者激活可导致炎症因子进一步表达,可能是糖尿病时血糖波动导致心肌细胞损害的主要病理生理机制之一。
[1] Sharma V,McNeill JH.Diabetic cardiomyopathy:where are we 40 years later?Can JCardiol,2006,22:305-308.
[2] Shichiri M,Kishikawa H,Ohkubo Y,etal.Long-term results of the kumamoto study on optimal diabetes control in type 2 diabetic patients.Diabetes Care,2000,23(Suppl.2):B21-B29.
[3] Keith D Brown,Estefania Claudio,Ulrich Siebenlist.The roles of the classical and alternative nuclear factor-B pathways: potential implications for autoimmunity and rheumatoid arthritis.Arthritis Research& Therapy,2008,10:212-226.
[4] 何 冰,赵 晟,张 巍,等.水杨酸钠对大鼠脂肪组织炎症因子表达的影响,陕西医学杂志,2009,3:267-269.
[5] Osorio-Fuentealba C,Valdes JA,Riquelme D,etal.Hypoxia stimulates via separate pathways erk phosphorylation and NF-κappaB activation in skeletal muscle cells in primary culture.JAppl Physiol,2009,29:[Epub ahead of print.
[6] Wang F,Kaur S,Cavin LG,etal.Nuclear-factorkappaB(NF-κappaB)and radical oxygen species play contrary roles in transforming growth factor-beta1(TGF-beta1)-induced apoptosis in hepatocellular carcinoma(HCC)cells.Biochem Biophys Res Commun,2008,377(4):1107-12.
[7] Annika Linde,Derek Mosier,Frank Blech,etal.Innate immunity and inflammation-new f rontiers in comparative cardiovascular patholμgy.Cardiovascular Research,2007,73:26-36.
[8] Jones WK,Brown M,Ren X,etal.NF-κappaB as an integrator of diverse signaling pathways: the heart of myocardial signaling?Cardiovasc Toxicol,2003,3:229-254.
[9] Sakon S,Xue X,Takekawa M,etal.NF-κBinhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death.EMBO J,2003,22:3898-3909.
[10] Ventura JJ,Cogswell P,Flavell RA,etal.JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species,2004,18:2905-2915.
[11] Pham CG,Bubici C,Zazzeroni F,etal.Ferritin heavy chain upregulation by NF-κB inhibits TNFa-induced apoptosis by suppressing reactive oxygen species,Cell,2004,119:529-542.
[12] Kamata H,Honda S,Maeda S,etal.Reactive oxygen species promote TNFa-induced death and sustained JNK activation by inhibiting MAPkinase phosphatases.2005,120:649-661.
猜你喜欢
现代仪器与医疗(2021年6期)2022-01-18
自我保健(2021年4期)2021-06-16
医学食疗与健康(2021年27期)2021-05-13
经济与管理(2020年4期)2020-12-28
恋爱婚姻家庭·养生版(2020年3期)2020-04-13
新闻传播(2018年13期)2018-08-29
家庭医学(2017年11期)2017-12-20
中国卫生标准管理(2015年15期)2016-01-15
中国医学装备(2015年10期)2015-12-29
中国当代医药(2015年23期)2015-03-01
