
三种Au(111)催化水煤气变换反应机理的比较
2010-11-06 07:01刘晓明倪哲明毛江洪王巧巧
物理化学学报 2010年6期
刘晓明 倪哲明 姚 萍 胥 倩 毛江洪 王巧巧
(浙江工业大学化学工程与材料学院,先进催化材料实验室,杭州 310032)
三种Au(111)催化水煤气变换反应机理的比较
刘晓明 倪哲明*姚 萍 胥 倩 毛江洪 王巧巧
(浙江工业大学化学工程与材料学院,先进催化材料实验室,杭州 310032)
采用密度泛函理论对三种水煤气变换反应(WGSR)机理(氧化还原机理、羧基机理、甲酸基的生成机理)在Au(111)面上的反应历程进行详细讨论.通过对表面吸附物种(H2O、CO、OH、O、H、CO2、COOH、HCOO)的吸附行为进行研究,得到最佳活性吸附中心.对三种机理中的14个基元反应的活化能进行分析,得出WGSR在Au(111)上按照羧基机理和氧化还原机理进行的可能性较大,按照甲酸基的生成机理进行的可能性较小.相比较羧基机理和氧化还原机理,反应更有可能按照羧基机理进行,最佳反应途径为H2O→-HOH→+COCOOH→+OHCO2.
密度泛函理论;Au(111)表面;反应机理
水煤气变换反应(CO+H2O=CO2+H2,简称WGSR)广泛应用在制氢工业、合成氨工业、甲醇合成、甲醇重整制氢中CO的去除、汽车尾气的净化处理以及燃料电池电动车等方面.因此,近年来水煤气变换反应引起国内外研究者的极大兴趣,随之,新颖的WGSR催化剂的研究也成为热点.目前在众多金属催化剂中,最令人感兴趣的是纳米金催化剂.纳米金可以单独作为催化剂也可以负载不同载体成为负载型金催化剂,并且都具有良好的催化效果.1989年Haruta等[1]对金纳米颗粒在低温下对CO的催化反应进行了研究,发现当尺寸小于10 nm的金颗粒分散到氧化物载体上时,对CO氧化表现出很高的催化活性.Andreeva等[2]比较了Au/Fe2O3和纯Fe2O3催化剂在水煤气变换反应中的活性,结果表明在240-360℃范围内,Au/Fe2O3的催化活性比纯Fe2O3的高.
为什么Au纳米颗粒在WGSR中具有较高催化活性,这就需要更好地了解在WGSR中纳米Au的催化机理.关于催化机理,人们曾对Fe、Ni等催化WGSR中的表面吸附物进行了大量的研究[3-5].最近Mavrikakis等[6]用密度泛函理论研究Pt催化WGSR反应机理,得到反应最有可能按照羧基机理进行. Gokhale等[7]用密度泛函理论研究了低温下Cu催化WGSR反应机理,提出羧基机理为主要反应机理.那么Au催化WGSR是否也按照羧基机理[6-7]进行?是否有可能按照氧化还原机理[8-12]或者甲酸基的生成机理[13-15]进行?Liu等[16]曾用密度泛函理论对Au(100)表面催化WGSR的反应机理进行了研究,提出反应有可能按照生成羧基中间体的机理进行.但是目前,人们对纳米Au在WGSR中的研究大都局限在O、H、OH、H2O、CO、CO2以及HOCO表面物种的反应[17-19]上,对整个反应历程和反应机理的研究不多,特别是对甲酸基的生成机理的实验研究更是少之又少.
由于实验的复杂性和表征手段的局限性,要从实验方面去解释纳米Au上WGSR反应机理具有很大难度.因此本文构建Au(111)表面的周期性平板模型,采用密度泛函理论,对Au(111)表面上8个吸附物种(H2O、CO、OH、O、H、CO2、COOH、HCOO)的吸附行为进行系统研究,得到最佳活性吸附中心.对文献中提出的三种WGSR反应机理进行详细讨论,分别模拟三种机理在Au(111)面上的反应历程,通过对反应活化能的分析,得出最佳反应途径,希望为Au催化WGSR的实验研究提供理论基础和参考.
1 计算模型和方法
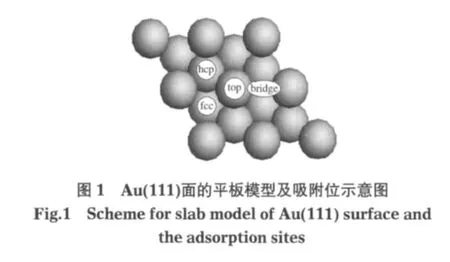
目前,常采用金属簇模型对金属催化剂进行模拟.这种计算方法的可信度依赖于选取簇的大小和簇模型体系的正确性.但在计算中很难找到与真实结构完全相符的簇模型,因此,计算值与真实值会存在一定偏差.周期性结构能够充分考虑到周围原子的影响,从而有效地减少这种偏差,因此本文采用周期性结构.本文分别选取三层平板和四层平板来模拟该表面,计算结果表明所选取层数不同对吸附能稍有影响,但是吸附物质的几何构型和吸附位的稳定顺序不变,考虑到计算精度和计算效率,本文选取三层平板厚度,采用(2×2)超晶胞模型来模拟Au(111)表面(覆盖率1/4).相邻两层平板间的真空层厚度为1 nm,以确保平板间的相互作用足够小. Au(111)表面的吸附位置一般有四种:顶位(top)、桥位(bridge)、面心立方位(fcc)和六角密积(hcp).将Au (111)平板模型和吸附位置示于图1.
本文的计算采用Materials Studio 4.3软件包中的Dmol3模块[20].计算中对内层电子作冻芯处理,采用有效核势(ECP)的赝势;价电子波函数采用双数值基组加极化函数(DNP)展开;交换相关势采用广义梯度积分和Perdew-Wang-91泛函相结合的方法(GGAPW91)[21-22];Brillouinzone积分的Monkhorst-Pack网络参数设置为4×4×1,Methfessel-Paxton smearing设为0.005 Ha;计算过程中精度设为fine(能量差异小于10-5Ha,每个原子上的力低于2×10-3Ha·eV-1,原子位移小于5.0×10-4nm),优化收敛精度取程序内定值;采用上述方法对Au晶胞参数进行优化,得到a为0.4170 nm,与实验值0.4078 nm[23]相近.另外本课题组[24]曾采用相同的平板模型研究了Cu催化WGSR的反应机理,结果与实验值符合较好.这说明本文采用的模型和计算方法是合理的.
Nichols等[25]曾对洁净的Au(111)进行X射线研究,实验结果表明其表面有1.3%的扩张,吸附能计算结果表明,与表层原子自由放开进行优化相比较,固定底物优化时,体系的总能量略有升高,但各种吸附位的稳定顺序和吸附物质的几何构型没有变化.因此在本文在构型优化时底物全部固定.根据文献报道[26],自旋极化对吸附构型影响很小,所以我们在构型优化时选用自旋极化不受限制.采用上述方法对各基元反应的反应物和产物进行结构优化,将优化后得到的稳定构型分别作为反应的初态和终态,采用完全线性同步和二次同步变换LST/QST方法[27]搜索各基元反应的过渡态.
2 结果与讨论
对文献中报道的三种WGSR机理进行比较,具体反应步骤列于表1.由表1可以看出三种机理主要区别在于第(4)-(6)步,计算中将着重讨论这三个步骤.第(1)、(2)和(7)步不做讨论.
2.1 各物种在Au(111)表面上的吸附
吸附能(EB)的定义是指吸附前后各物质总能量的变化,其符号大小可以表示发生吸附的可能性和吸附的程度.本文根据计算模型将EB定义为EB=E(A/surface)-(EA+Esurface).其中E(A/surface)表示A分子吸附Au(111)表面时体系的总能量,EA表示A分子的能量,Esurface表示Au(111)面的能量.
在三种反应机理中共有8个吸附物种(H2O、CO、OH、O、H、CO2、COOH、HCOO),本文对这些物种在Au(111)面所有吸附位上的EB进行计算,通过EB的比较得到最佳吸附中心.将计算得到各物种稳定吸附构型的结合能及结构参数列于表2.并将各个物种的最佳吸附位示于图2.
由表2和图2可知,H和O原子最佳吸附位都为面心立方(fcc)位,如图2(a,b)所示;CO的最佳吸附位为fcc,通过C端垂直地与Au表面作用,如图2(c)所示;OH自由基最佳吸附位为桥(bridge)位,以O为接触端倾斜地吸附在Au表面,如图2(d);H2O分子最佳吸附位为顶(top)位,以O为接触端平行地吸附在Au表面,如图2(e);CO2的最佳吸附位为bridge位,以C为接触端平行地吸附在Au表面,如图2(f)所示.
羧基COOH在Au(111)表面有两种稳定构型,分别标记为反式羧基(trans-COOH)、顺式羧基(cis-COOH),稳定构型如图2(g,h).其中O—H键中H原子靠近Au(111)表面的构型称为trans-COOH,θCOH和θOCO分别为110.2°和123.4°,表面吸附能为-160.2 kJ·mol-1,trans-COOH中OH的振动频率为3713 cm-1;另一种O—H键中H原子远离Au(111)表面的构型称为cis-COOH,θCOH和θOCO分别为107.4°和124.3°,表面吸附能为-154.4 kJ·mol-1,cis-COOH中OH的振动频率为3746 cm-1.由于trans-COOH的结构在Au(111)表面比cis-COOH稳定(结合能低5.8 kJ· mol-1),因此本文选用trans-COOH作为反应的中间体.

表1 Au(111)催化WGSR的氧化还原机理、羧基机理、甲酸基的生成机理Table 1 Redox mechanism,carboxyl mechanism,and formate intermediate mechanism for WGSR on Au(111)

表2 WGSR反应物种在Au(111)表面上的最佳吸附位、几何结构参数及表面结合能Table 2 Preferred sites,WGSR geometric parameters,adsorption energies for WGSR species on Au(111)surface

甲酸根HCOO与羧基COOH为同分异构体,其在Au(111)表面上也有两种稳定构型,分别标记为双接触甲酸根(di-HCOO)、单接触甲酸根(mono-HCOO),稳定构型如图2(i)、图2(j).其中以两个O为接触点与表面发生作用的构型称为di-HCOO,最佳吸附位为top-top位,θOCO和θOCH分别为131.5°、113.8°,Au—O的距离为0.2333 nm,表面结合能为-183.4 kJ·mol-1.另一种以一个O为接触点与Au表面发生作用的构型称为mono-HCOO,最佳吸附位为hcp位,θOCO和θOCH分别为130.4°、114.4°,Au—O的距离为0.2330 nm,表面结合能为-165.0 kJ·mol-1. HCOO上C—H的振动频率为3040 cm-1.由于di-HCOO的结构在Au(111)表面比mono-HCOO稳定(结合能低18.4 kJ·mol-1),因此本文选用di-HCOO作为反应的中间体.

2.2 三种反应机理
将三种可能反应机理中主要基元反应的活化能Ea和反应焓ΔH列于表3.
2.2.1 氧化还原机理
将氧化还原机理中的第(3)-(6)步在Au(111)上反应的初始态(IS)、过渡态(TS)、终态(FS)列于图3.
H2O→OH+H(R3).初始态(IS)中H2O分子平行吸附于Au(111)面的top位,解离时,H2O的一个O—H键断裂,其中一个H向临近的top位迁移,而未断裂的O—H向bridge迁移,这时反应过程处于过渡态(TS),H与O—H的距离为0.2754 nm,O—H的键长为0.0983 nm,θHOH为98.7°.最后H和O—H各自稳定吸附在fcc位和bridge位,达到终态(FS),如图3R(3)所示.比较初态和终态的总能量,发现该步骤反应前后的ΔH为115.8 kJ·mol-1,为吸热反应.反应的活化能垒Ea为191.1kJ·mol-1.

表3 各基元反应在Au(111)面的活化能Eα和反应焓ΔHTable 3 Activation barriers(Eα)and enthalpy changes(ΔH)of elementary reactions on Au(111)surface
OH→O+H(R4).初始态中,OH以O端为吸附点倾斜地吸附在Au(111)表面的bridge位,解离过程如图3R(4),在过渡态中,O—H键拉长的同时,H向邻近的hcp位移动,H—O的键长为0.1544 nm.最后O—H键断裂,O和H分别吸附在邻近的fcc位,到达终态.该步骤反应前后ΔH为134.1 kJ·mol-1,为吸热反应.反应的活化能垒Ea为166.0 kJ·mol-1.
OH+OH→H2O+O(R5).两个OH歧化反应产生H2O,首先我们构建OH在Au(111)表面的共吸附模型,并对其优化,发现其中一个OH由原来的bridge位向top位转移,另一个仍留在bridge位.以此作为该基元反应的初始态,进行过渡态搜索,过程如图3R(5).在过渡态中,其中bridge位上的OH键拉长并发生断裂,断裂后H向另一个在top位的OH移动,结合生成H2O,两个O—H的键长分别为0.1960和0.1395 nm,两个O之间的距离为0.2438 nm.达到终态时,OH键断裂后的O稳定吸附在hcp位, H2O稳定吸附在top位.该步骤反应前后ΔH为18.3 kJ·mol-1,为吸热反应.反应的活化能垒Ea为42.4 kJ·mol-1.
CO+O→CO2(R6).将CO和O在Au(111)表面上的共吸附模型进行优化,发现CO由原来的fcc位迁移到临近的top位,O未发生变动依然留在fcc位,我们将优化后的共吸附模型作为反应过程的初始态,解离过程如图3R(6).在搜索过渡态过程中,O开始由fcc位向bridge位发生迁移并向CO靠近, CO仍留在top位但是发生了小角度的倾斜,两个C—O的键长分别为0.1245和0.2101 nm.最后CO和O结合生成CO2,在终态时,CO2稳定吸附在bridge位.该步骤反应前后ΔH为-53.1 kJ·mol-1,为放热反应.反应的活化能垒Ea为55.9 kJ·mol-1.
2.2.2 羧基机理
将羧基机理中的第(4)-(6)步在Au(111)上反应的初始态(IS)、过渡态(TS)、终态(FS)列于图4.
CO+OH→COOH(C4).将CO和OH在Au(111)表面上的共吸附模型进行优化,发现CO由原来的fcc位迁移到临近的top位,OH未发生变动依然留在brigde位,我们将优化后的共吸附模型作为反应过程的初始态,解离过程如图4C(4).在搜索过渡态过程中,CO倾斜并由原来的top位向临近的bridge位迁移,OH逐渐向CO靠近,C—O和O—H的键长分别为0.1177和0.0969 nm,C—O与O—H的距离为0.2261 nm.最后OH的O与CO的C结合生成trans-COOH,trans-COOH稳定地吸附在top位.该步骤反应前后ΔH为-15.4 kJ·mol-1,为放热反应.反应的活化能垒Ea为42.4 kJ·mol-1.
COOH→CO2+H(C5).初始态中,trans-COOH以C端为吸附点吸附在top位,以此作为反应的起点,搜索过渡态,解离过程如图4C(5).通过对过渡态分析后发现,H从COOH中脱离出来并向fcc位移动, H脱离之后的CO2整体向临近的bridge位移动,H与CO2的两个O的距离分别为0.1258和0.1666 nm.最后H和CO2分别稳定地吸附在fcc位和bridge位.该步骤反应前后ΔH为-39.5 kJ·mol-1,为放热反应.反应的活化能垒Ea为84.9 kJ·mol-1.

COOH+OH→CO2+H2O(C6).将trans-COOH和OH的共吸附模型进行优化,发现OH由原来的bridge位向fcc靠近,trans-COOH依然留在top位,我们将此时的共吸附模型作为反应的初始模型搜索过渡态,解离过程如图4C(6).在过渡态中trans-COOH的O—H键拉长且H向OH靠近,H逐渐从trans-COOH中脱离与OH生成H2O,H2O的两个O—H键长分别为0.0981和0.1007 nm,CO2中两个C—O键长分别为0.1366和0.1188 nm.最后生成的CO2和H2O分别稳定地吸附在bridge位和top位.该步骤反应前后ΔH为-19.3 kJ·mol-1,为放热反应.反应的活化能垒Ea为37.6 kJ·mol-1.
将甲酸基的生成机理中的第(4)-(6)步在Au(111)上反应的初始态(IS)、过渡态(TS)、终态(FS)列于图5.
CO2+H→HCOO(F4).将CO2和H的共吸附模型进行优化,发现H由原来的fcc位向临近的hcp移动,CO2留在bridge位,将此作为反应的起点搜索过渡态,解离过程如图5F(4).发现H向CO2的C端靠近,H与CO2的距离为0.1671 nm,两个C—O的键长分别为0.1178 nm.最后C—H连接成键生成HCOO,终态时HCOO稳定地吸附在top-top位.该步骤反应前后ΔH为-72.4 kJ·mol-1,为放热反应.反应的活化能垒Ea为137.0 kJ·mol-1.
HCOO+O→CO2+OH(F5).将HCOO和O的共吸附模型进行优化,发现O由fcc位向top位迁移, HCOO依然吸附在top-top位,但是HCOO向O发生翻转,将此作为反应的起点搜索过渡态,解离过程如图5F(5).发现HCOO上的C—H键逐渐拉长并且H向O靠近,最后C—H键断裂,H与O成键生成OH,CO2中的两个C—O键长分别为0.1194和0.1326 nm,O—H的键长为0.1796 nm.反应终态时生成的CO2和OH各自稳定地吸附在表面的bridge位.该步骤反应前后ΔH为-61.7 kJ·mol-1,为放热反应.反应的活化能垒Ea为84.9 kJ·mol-1.
HCOO+OH→CO2+H2O(F6).将HCOO和OH的共吸附模型进行优化,发现OH由bridge位向临近的fcc移动,HCOO的H向下翻转,将此作为反应的起点搜索过渡态,解离过程如图5F(6).在搜索过渡态过程中HCOO的C—H拉长并且H慢慢向OH靠近,最后H从HCOO上脱离与OH生成H2O, H2O中的两个O—H键长分别为0.9976和0.9630 nm,CO2中的两个 C—O键长分别为 0.1171和0.1175 nm.终态时H2O与CO2分别稳定吸附在top和bridge位.该步骤反应前后ΔH为-43.4 kJ·mol-1,为放热反应.反应的活化能垒Ea为59.8 kJ·mol-1.


2.2.4 三种机理的比较
对三种反应机理在Au(111)上的最佳反应途径进行比较,将各个反应的活化能垒示于图6.
由图6可看到,反应按照甲酸基的生成机理进行,需要经过OH分解反应、CO的氧化和HCOO的生成三步反应,且这三步反应的活化能都非常高,所以反应按照甲酸基的生成机理进行的可能性很小.比较氧化还原机理和羧基机理发现,主要不同点是:氧化还原机理经过OH歧化反应和CO氧化两步反应,活化能分别为42.4和55.9 kJ·mol-1;羧基机理经过中间体COOH生成和COOH与OH歧化反应两步反应,活化能分别为42.4和37.6 kJ·mol-1.说明反应既有可能按照氧化还原机理进行,也有可能按照羧基机理进行,但由于COOH与OH歧化反应的活化能只有37.6 kJ·mol-1,所以按照羧基机理进行的可能性更大,并且最佳反应途径为另外,从反应热来看此途径,OH歧化反应的ΔH为18.3 kJ·mol-1,而生成COOH反应的ΔH为-15.4 kJ·mol-1,说明在低温条件下更有利于COOH的生成反应.综上所述,在Au(111)面上WGSR按照羧基机理反应的可能性较大,其次是氧化还原机理,而按照甲酸基的生成机理反应的可能性较小.
3 结 论
采用密度泛函理论对Au(111)上水煤气变换反应的三种可能反应机理进行了研究,对各个吸附物种在Au(111)面的吸附构型进行了几何优化进而得
1 Haruta,M.;Yamada,N.;Kobayashi,T.;Iijima,S.J.Catal.,1989, 115:301
2 Andreeva,D.;Idakiev,V.;Tabakova,T.J.Catal.,1996,158:354
3 Grenoble,D.C.;Estadt,M.M.;Ollis,D.F.J.Catal.,1981,67:90
4 Jager,B.;Espinoza,R.Catal.Today,1995,23:17
5 Campbell,C.T.;Ernst,K.H.ACS Symp.Ser.,1992,482:130
6 Grabow,L.C.;Gokhale,A.A.;Evans,S.T.;Dumesic,J.A.; Mavrikakis,M.J.Phys.Chem.C,2008,112:4608
7 Gokhale,A.A.;Dumesic,J.A.;Mavrikakis,M.J.Am.Chem.Soc., 2008,130:1402
8 Ovesen,C.V.;Clausen,B.S.;Hammershoi,B.S.;Steffensen,G.; Askgaard,T.;Chorkendorff,I.;Nørskov,J.K.;Rasmussen,P.B.; Stoltze,P.;Taylor,P.J.Catal.,1996,158:170
9 Nakamura,J.;Cambell,J.M.;Campbell,C.T.J.Chem.Soc., 1990,86:2725到稳定构型,对14个基元反应的反应物、反应中间体、过渡态和产物进行研究,得到了各个基元反应所需的活化能,得到以下结论:
(1)WGSR在Au(111)上按照氧化还原机理进行的最佳途径为但低温下不利于按此途径进行;按照羧基机理进行的最佳途径为并且低温下反应也可按照此途径进行.按甲酸基的生成机理进行的最佳途径为并且低温下反应也有可能按照此途径进行.
(2)纵观整个反应历程,从能量学角度上看, WGSR在Au(111)上按照羧基机理和氧化还原机理进行的可能性较大,按照甲酸基的生成机理进行的可能性较小.并且相比较羧基机理和氧化还原机理,由于COOH与OH歧化反应的活化能只有37.6 kJ· mol-1,所以反应更有可能按照羧基机理进行,且最佳反应途径为
10 Bunluesin,T.;Gorte,R.J.;Graham,G.W.Appl.Catal.,1998,15: 107
11 Koryabkina,N.A.;Phatak,A.A.;Ruettinger,W.F.;Farruto,R.J.; Rebeiro,F.H.J.Catal.,2003,217:233
12 Rhodes,C.;Hutchags,G.J.;Wand,A.M.Catal.Today,1995,23: 43
13 Herwijnen,T.V.;Jong,W.A.D.J.Catal.,1980,63:94
14 Campbell,C.T.;Koel,B.E.;Daube,K.A.J.Vac.Sci.Technol., 1987,5:810
15 Yoshihara,J.;Parker,S.;Schafer,A.;Campbell,C.T.Catal.Lett., 1995,31:313
16 Liu,P.;Rodriguez,J.A.J.Chem.Phys.,2007,126:164705
17 Gong,J.L.;Ojifinni,R.A.;Kim,T.S.;Stiehl,J.D.;McClure,S. M.;White,J.M.;Mullins,C.B.Top.Catal.,2007,4:57
18 Shubina,T.E.;Hartnig,C.;Koper,M.T.M.Phys.Chem.Chem. Phys.,2004,6:4215
19 Phatak,A.A.;Delgass,W.N.;Ribeiro,F.H.;Schneider,W.F. J.Phys.Chem.C,2009,113:7269
20 Materials Studio.Version 4.3.San Diego:Accelrys Inc.,2008
21 Perdew,J.P.;Chevary,J.A.;Vosko,S.H.;Jackson,K.A.; Pederson,M.R.;Singh,D.J.;Fiolhais,C.Phys.Rev.B,1992,46: 6671
22 White,J.A.;Bird,D.M.;Payne,M.C.;Stich,I.Phys.Rev.Lett., 1994,73:1404
23 Mai,S.W.;Zhou,G.D.;Li,W.J.Advanced inorganic structural chemistry.Beijing:Peking University Press,2001:302 [麦松威,周公度,李伟基.高等无机结构化学.北京:北京大学出版社, 2001:302]
24 Mao,J.H.;Ni,Z.M.;Pan,G.X.;Xu,Q.Acta Phys.-Chim.Sin., 2008,24:2059 [毛江洪,倪哲明,潘国祥,胥 倩.物理化学学报,2008,24:2059]
25 Nichols,R.J.;Nouar,T.;Lucas,C.A.;Haiss,W.;Hofer,W.A. Surf.Sci.,2002,513:263
26 Ge,Q.;Jenkins,S.J.;King,D.A.Chem.Phys.Lett.,2000,327: 125
27 Zhang,X.H.;Zhang,M.H.;Jiang,H.X.J.Mol.Catal.,2006,20: 535 [张霄航,张敏华,姜浩锡.分子催化,2006,20:535]
Comparison of Three Reaction Mechanisms for the Water Gas Shift Reaction on Au(111)Surface
LIU Xiao-Ming NI Zhe-Ming*YAO Ping XU Qian MAO Jiang-Hong WANG Qiao-Qiao
(Laboratory of Advanced Catalytic Materials,College of Chemical Engineering and Materials Science, Zhejiang University of Technology,Hangzhou 310032,P.R.China)
A detailed density functional theory(DFT)investigation revealed three possible mechanisms(redox mechanism,carboxyl mechanism,and formate intermediate mechanism)for the water-gas shift reaction on Au(111) surface.All the pertinent species(H2O,CO,OH,O,H,CO2,COOH,HCOO)were calculated.We obtained their preferred adsorption sites.We characterized the reaction pathway containing 14 elementary steps and calculated the reaction potential energy surfaces.The calculation results show that the carboxyl mechanism and the redox mechanism are feasible while the formate intermediate mechanism is unlikely because of its high formation barrier.Our calculations also show that the carboxyl mechanism is more probable compared with the redox mechanism and the most feasible reaction pathway is H2O→-HOH→+COCOOH→+OHCO2.
Density functional theory;Au(111)surface;Reaction mechanism
O641
Received:January 15,2010;Revised:March 8,2010;Published on Web:April 30,2010.
*Corresponding author.Email:jchx@zjut.edu.cn;Tel:+86-571-88320373.
The project was supported by the Natural Science Foundation of Zhejiang Province,China(Y406069).
浙江省自然科学基金(Y406069)资助项目
ⒸEditorial office of Acta Physico-Chimica Sinica
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
应用化工(2014年10期)2014-08-16
应用化工(2014年7期)2014-08-09

- 物理化学学报的其它文章
- 化学气相沉积法合成Fe/CMK-5及其对溶菌素的吸附性能