
有机锡氧簇合物[(μ-O)(μ-OH)Sn2(n-Bu)4]2(C5H4NCO2)2的合成、结构和量子化学研究
2010-09-15 11:44邝代治冯泳兰张复兴王剑秋
无机化学学报 2010年12期
邝代治冯泳兰 张复兴 王剑秋
(功能金属有机材料湖南省普通高等学校重点实验室,衡阳师范学院化学与材料科学系,衡阳 421008)
有机锡氧簇合物[(μ-O)(μ-OH)Sn2(n-Bu)4]2(C5H4NCO2)2的合成、结构和量子化学研究
邝代治*冯泳兰 张复兴 王剑秋
(功能金属有机材料湖南省普通高等学校重点实验室,衡阳师范学院化学与材料科学系,衡阳 421008)
吡啶-4-甲酸与(n-Bu)2SnO 反应合成二(μ-氧)二(μ-羟基氧)桥联四[二(正丁基)锡]二(吡啶-4-甲酸酯),经元素分析、1H NMR、IR 和 X-射线衍射表征分子结构,该配合物晶体属单斜晶系,空间群 P21/n,晶胞参数:a=1.3065(3)nm,b=1.3145(2)nm,c=1.6608(3)nm,β=96.180(5)°,V=2.8357(9)nm3,Z=4,Dc=1.454 g·cm-3,μ(Mo Kα)=1.784 mm-1,F(000)=1248,R1=0.0449,wR2=0.1241(I>2σ(I))和R1=0.0887,wR2=0.1756(对所有的衍射)。共收集 15802 个衍射点,其中独立衍射点 4 885 个,可观察衍射(I>2σ(I))点 2733 个用于结构精修。中心Sn原子形成五配位畸变三角双锥构型,分子间通过N…H-O氢键作用形成二维网络结构。利用量子化学G98W软件,在Lanl2dz基组对配合物的稳定性、前沿分子轨道组成及能量进行研究。
有机锡氧化合物;合成;晶体结构;量子化学
自从Chandrasekhar等报道六聚鼓形有机锡氧化合物后[1],引起许多学者的兴趣[2],相继合成出具有环形、笼形锡氧簇的有机锡化合物。近年来,通过烃基锡氧化合物与质子酸(如n-BuSn(O)-(OH)、羧酸和磷酸)的反应合成双氧帽有机锡簇合物[3];Chandrasekhar还报道了 H3PO3存在下 n-BuSn(O)(OH)与 p-XC6H4OH(X:H,Cl,Br 和 I)反应[4],合成了一些含Sn-O结构的梯形、鼓形、立方形、帽形和足球形等新颖的锡氧簇化合物[5-6],我们最近也合成了含Sn2O2结构的新有机锡化合物[7],本文报道含有O-Sn和C-Sn键并具有Sn2O2骨架结构的四核环状梯形有机锡氧簇合物[(μ-O)(μ-OH)Sn2(n-Bu)4]2(C5H4N CO2)2,并在Lanl2dz基组水平研究标题配合物的稳定性、前沿分子轨道组成及能量。
1 实验部分
1.1 试剂和仪器
所有试剂均为化学纯。IR用日本岛津FTIR-8700 红外光谱仪(4000~400 cm-1,KBr压片) 测定;1H NMR用Bruker Av400核磁共振仪 (TMS内标,DCCl3溶剂)测定;元素分析用PE-2400(Ⅱ)元素分析仪测定;晶体结构用SMART APEXⅡ型X射线衍射仪(德国布鲁克公司)测定。
1.2 配合物的合成
取 0.2489 g(1 mmol)(n-Bu)2SnO 和 0.0615 g(0.5 mmol)吡啶-4-甲酸于30 mL乙醇溶液中,搅拌混合后,回流反应10 h,冷却,旋转蒸发除去部分溶剂,低温放置数天,析出无色晶体0.16 g,收率为51.6%,m.p.:138~139 ℃。元素分析 C44H82N2O8Sn4,Mr=1 241.97,计算值(%):C,42.55;H,6.65;N,2.26。实测值(%):C,42.75;H,6.60;N,2.18。1H NMR(DCCl3,ppm)δ:0.90~1.72(m,8×9H,n-Bu),7.78~8.72(m,2×4H,hydrogen of pyridinyl)。IR(KBr):2 956.7(vs),2 923.9(vs),2 869.9(s),2856.4(m),1645.2(vs),1602.7(w),1558.4(m),1463.9(w),1407.9(m),1348.1(vs),1311.5(m),1290.3(w),1 251.7(w),1 226.6(w),1 141.8(w),1 060.8(w),1 001.0(m),962.4(w),862.1(w),846.7(w),767.6(s),709.8(m),682.8(s),624.9(s),582.5(s),549.7(m),435.9(w)cm-1.
1.3 晶体结构分析
选取一颗 0.18 mm×0.15 mm×0.10 mm 的晶体,采用石墨单色化的 Mo Kα 射线 (λ=0.071073 nm),于293(2)K,以φ~ω扫描方式,在X-射线衍射仪上收集衍射数据。在 1.98°≤θ≤25.05°范围内,在 h=-15~15,k=-15~15,l=-19~15 范围内收集 15802 个数据,其中独立衍射点 4885 个(Rint=0.0321),I>2σ(I)的可观察衍射为2733个,晶体结构由直接法解出。对全部非氢原子及其各向异性热参数进行全矩阵最小二乘法修正,理论加氢法确定氢原子的位置坐标。最终收敛偏差因子为 R1=0.044 9,wR2=0.124 1(对 I>2σ(I)的衍射)和 R1=0.088 7,wR2=0.175 6(对所有的衍射);w=1/[σ2(Fo2)+(0.076 4P)2+3.637 3P],其中P=(Fo2+2Fc2)/3,S=1.125,(Δ/σ)max=0.000,差值 Fourier图最高和最低残余电子密度峰 (Δρ)max=644 e·nm-3,(Δρ)min=-401 e·nm-3。衍射数据收集用 Smart程序、衍射数据还原用Saint程序、结构解析用Shelxtl程序完成。
CCDC:715879。
2 结果与讨论
2.1 红外光谱
标题配合物的红外光谱特征峰较为明显。(1)在高频区出现丁基的特征吸收峰,分别为甲基(2956.7、2869.9 cm-1)和亚甲基(2923.9、2856.4 cm-1)的C-H伸缩振动吸收峰。(2)在1645.2 cm-1出现尖锐而特强的单峰,为吡啶甲酸与锡原子连接成有机锡羧酸酯后的羰基的反对称伸缩吸收峰,羰基的对称伸缩吸收峰在1348.1 cm-1,也是尖锐而特强的单峰,且证明配合物中只有一个这样的酯基存在,反对称与对称伸缩振动吸收峰波数差ΔⅤ(CO2)=297.1 cm-1,表明 Ⅴas(CO2)未明显地向低频区移,Ⅴs(CO2)未明显地向高频区移,羰基是以单氧形式与锡原子配位[8-9]。(3)红外光谱分别出现 435.9 和 549.7 cm-1弱吸收峰,表明分子有O-Sn和Sn-C键存在[10-11],与晶体分子结构相吻合。
2.2 晶体结构
配合物的主要键长和键角列于表1,分子结构和二维网络结构图分别见图1、2。从图1和键参数看出,配合物通过μ-氧和μ-羟基氧桥联 2个(二正丁基)锡形成Sn、O原子交替,含Sn2O2结构的四元环,2个这种四元环通过Sn、O原子交错二聚,形成具有3个这样的四元环稠合的梯形锡氧簇结构。在晶体分子结构中,Sn原子除与氧原子相链外,还分别连接着2个正丁基,并还有1个吡啶-4-甲酸根离子与其中Sn2原子配合,因此,标题配合物是含有C-Sn和O-Sn键的金属有机锡羧酸酯配合物。

表1 配合物的主要键长和键角Table 1 Selected of bond lengths(nm)and bond angles(°)of the complex
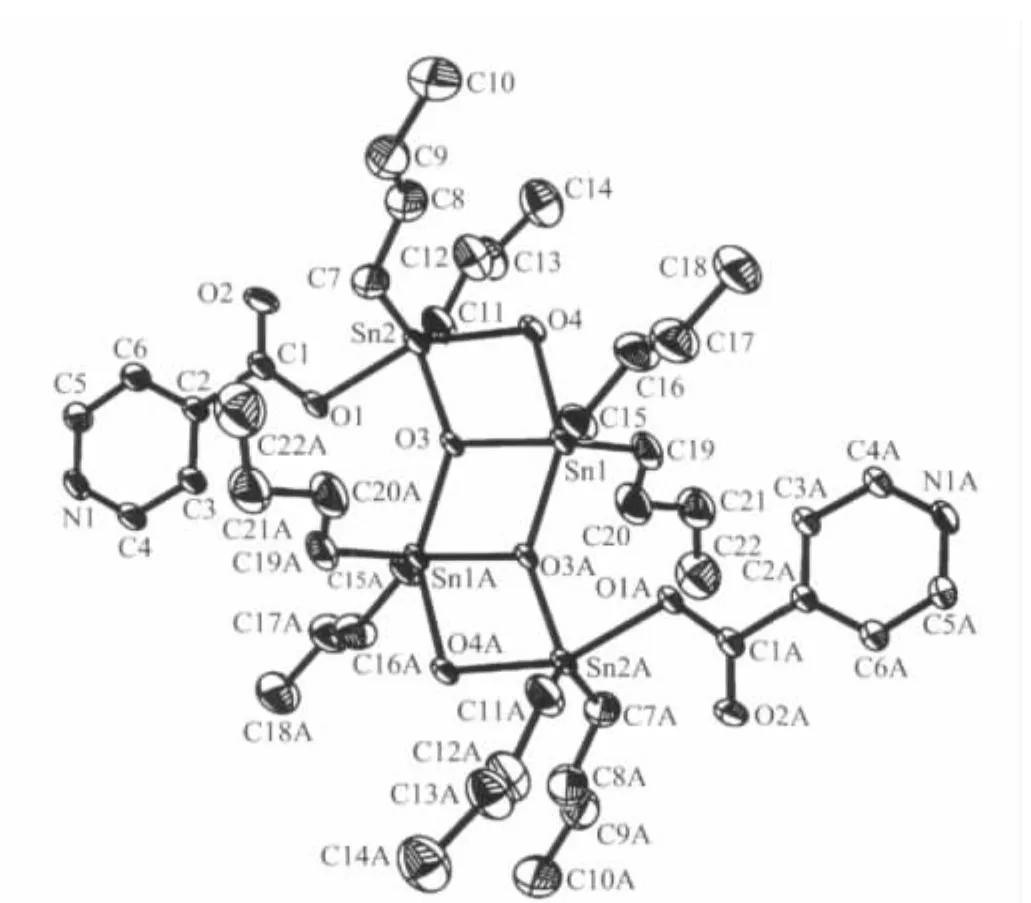
图1 配合物[(μ-O)(μ-OH)Sn2(n-Bu)4]2(C5H4NCO2)2的分子结构图Fig.1 Molecular structure of the complex at 15%probability thermal ellipsoids
在中心Sn原子周围,占据着赤道平面位置的氧原子几乎于同一平面,由于丁基的空间效应,占据了轴向位置的碳链向外偏,使∠C15-Sn1-C19键角和∠C7-Sn2-C11键角变小。
配合物中构成四元环骨架锡氧原子的Sn-O键有 3类:Sn1-O4 2.138(7)nm,Sn1-O3 2.046(5)nm,Sn1-O3A 2.118(5)nm,Sn2-O4 2.162(6)nm,Sn2-O3 2.004(5)nm,Sn2-O1 2.179(5)nm,均不相等,与梯形Sn2O2结构的Sn-O键相差不大[12];比类似锡氧簇合物的羧基氧原子形成的Sn-O键长[13]。四元环的二面角:∠O4-Sn2-O3-Sn1为 2.6(3)°,∠O3-Sn1-O3A-Sn1A=0.03(6)°,表明中间四元环的Sn2O2结构有较好的共平面性。从中心Sn原子分别相连的键长、键角均不相等表明,Sn原子为畸变六面体构型。
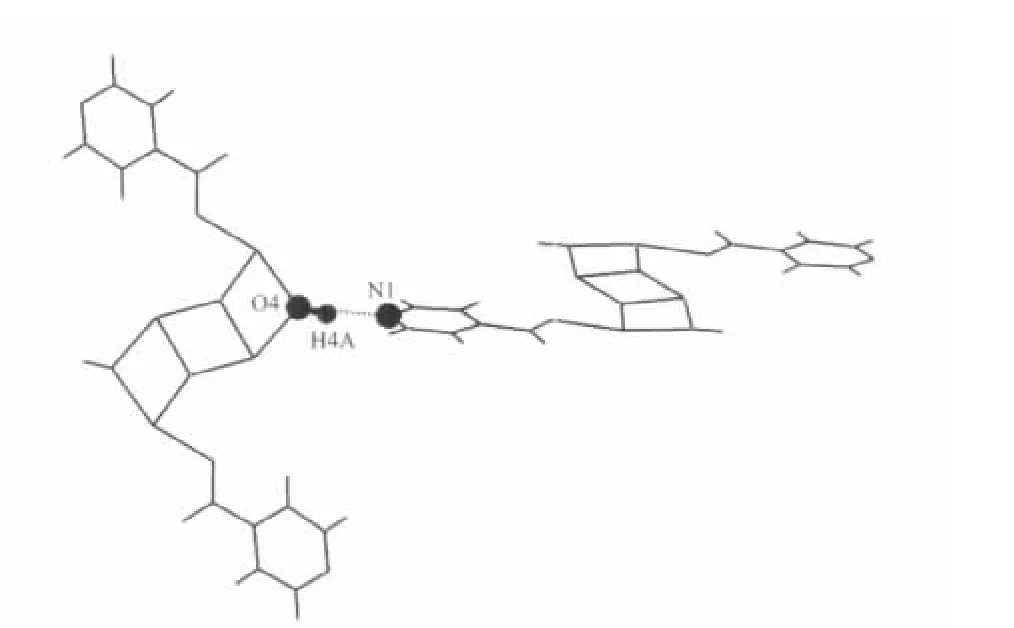
图2 配合物中N…H-O氢键作用结构图Fig.2 Hydrogen bonding interactions of the complex
吡啶的氮原子没有与锡原子发生配位,在晶体分子结构中,吡啶的氮N1原子与相邻分子的H4a原子之间形成氢键作用 (N1…H4a为0.187 7 nm,∠N1…H4a-O4为 156.2°),如图2,通过这种分子间的氢键作用,形成二维无限网状结构。
2.3 能量和分子轨道组成研究
根据晶体结构的各原子坐标位置,考虑到配合物的对称性,为节省计算机时,取其中一个结构单元用于Gaussian98W[14]程序的rhf/lanl2dz基组上,在P4计算机上进行量子化学单点计算。配合物计算涉及70原子,341个原子基函,863个初始高斯函数,110个α电子,109个β电子。
计算得到配合物体系量为-1216.5935662 a.u.,轨道的能量 HOMO为-0.326 38 a.u.,LUMO的能量为0.031 75 a.u.,两前沿轨道间的能量间隙为0.358135 a.u.。从体系能和前沿轨道的能量分析,配合物较难失去电子,其基态较稳定。
为探索配合物的成键特征,对配合物分子轨道进行系统分析,用参与组合的各类原子的轨道系数的平方和来表示该部分在分子轨道中的贡献,并经归一化。把配合物原子分为五部分:(a)Sn原子;(b)氧原子 O;(d)氮原子 N;(e)碳原子 C;(f)氢原子H。
从分子轨道成份图3、4可知,在前沿轨道及其附近轨道成份有较明显变化,限于篇幅,只讨论前沿分子轨道。
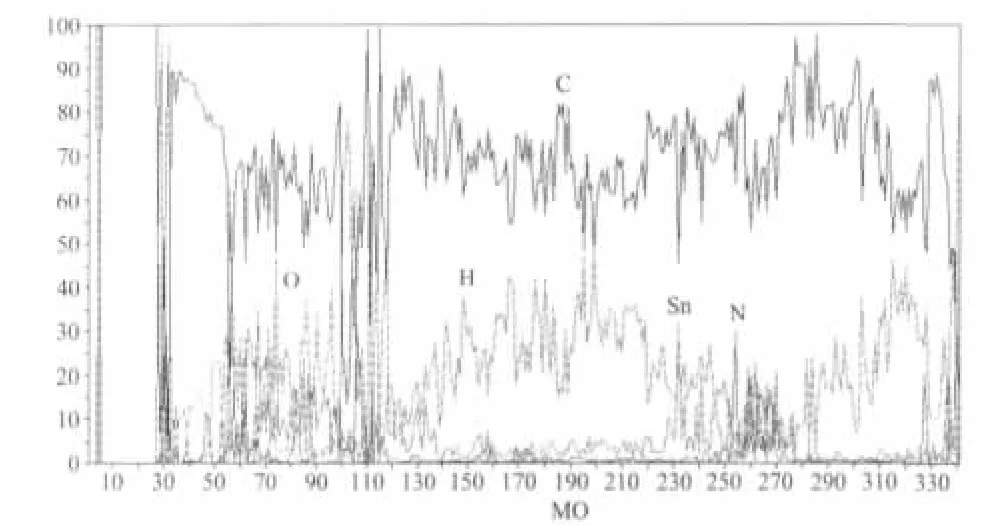
图3 配合物的分子轨道成份图Fig.3 Molecular orbitals composition of the complex at Lanl2dz level
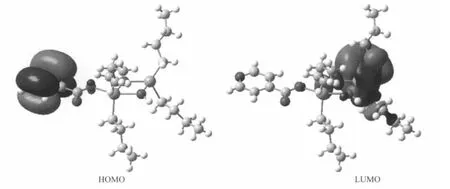
图4 配合物的前沿分子轨道示意图Fig.4 Schematic diagram of frontier MO for the complex
配合物中,碳原子对HOMO的贡献98.88%最大,其中吡啶环碳的贡献达98.63%,其他原子对HOMO的贡献很少,锡原子在HOMO成份0.01%,氧原子0.84%,氮原子0.18%,氢原子0.06%。
在最低未占分子轨道组成中,各原子对LUMO的贡献发生较明显变化,Sn原子对LUMO的贡献达60.88%,碳原子对LUMO的贡献32.05%(31.48%来自吡啶碳外的碳原子贡献),氧原子贡献为4.62%,氮原子的贡献0.01%,所有H原子的贡献2.43%。
比较HOMO与LUMO的轨道成份,发现配合物从基态向激发态电子转移时,主要是碳原子向Sn原子轨道转移,形成电荷转移配合物。
[1]Chandrasekhar V,Day R O,Holmes R R.Inorg.Chem.,1985,24:1970-1971
[2]Chandrasekhar V,Baskar V,Vittal J J.J.Am.Chem.Soc.,2003,125:2392-2393
[3]Ma C L,Han Y W,Zhang R F.Analytical Sciences,2007,23:x89-x90
[4]Chandrasekhar V,Baskar V,Gopal K.et al.J.Organometallics,2005,24:4926-4932
[5]ZHANG Chun-Hua(张春华),CHEN Man-Sheng(陈满生),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2005,21(9):1386-1388
[6]CHEN Man-Sheng(陈满生),KUANG Dai-Zhi(邝代治),ZHANG Chun-Hua(张春华),et al.Chinese J.Struct.Chem.(Jiegou Huaxue),2005,24(11):1249-1253
[7]WANGJian-Qiu(王剑秋),ZHANG Fu-Xiag(张复兴),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2007,23(5):871-874
[8]LUO Ning(罗 宁),SUN Li-Juan(孙丽娟),LIU Zhi-Zhong(刘治中),et al.Chinese J.Appl.Chem.(Yingyong Huaxue),2000,17(2):154-158
[9]XIE Qing-Lan(谢庆兰),XU Xiao-Hua(徐效华),ZHANG Doan-Kun(张殿坤).Acta Chimica Sinica(Huaxue Xuebao),1992,50:508-510
[10]ZHANG Fu-Xiag(张复兴),KUANG Dai-Zhi(邝代治),WANG Jian-Qiu(王剑秋),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2006,22(7):1321-1326
[11]Okawara R,Yasuda K.J.Organometallic Chem.,1964,1(4):356-359
[12]WANG Jian-Qiu(王剑秋),CHEN Zhi-Min(陈志敏),ZENG Rong-Ying(曾荣英),et al.Chinese J.Struct.Chem.(Jiegou Huaxue),2005,24(12):1387-1392
[13]YIN Han-Dong(尹汉东),WANG Chuan-Hua(王传华),MA Chun-Lin(马春林).Chinese J.Org.Chem.(Youji Huaxue),2003,23(5):470-474
[14]Aeleen F,Michael J F.Gaussian 98 User′s Reference,Gaussian,Inc.,Garnegie Office Park,Bldg.6 Pittaburgb,PA15106 USA.
Synthesis,Crystal Structure and Quantum Chemistry of the Organooxotin Cluster[(μ-O)(μ-OH)Sn2(n-Bu)4]2(C5H4NCO2)2
KUANG Dai-Zhi*FENG Yong-Lan ZHANG Fu-XingWANG Jian-Qiu
(Key Laboratory of Functional Organometallic Materials of College of Hunan Province,Department of Chemistry and Material Science,Hengyang Normal University,Hengyang,Hunan 421008)
A novel organooxotin complex of di-μ-oxygen and di-μ-hydroxyl bridged tetra(bisbutyltin)bis(pyridine-4-carboxylate)has been synthesized by the reaction of n-Bu2SnO with pyridine-4-carboxylic acid and characterized by IR,1H NMR spectra and elemental analysis.The crystal structure has been determined by X-ray diffraction.It crystallizes in the monoclinic,space group P21/n with a=1.3065(3)nm,b=1.3145(2)nm,c=1.6608(3)nm,β=96.180(5)°,V=2.835 7(9)nm3,Z=4,Dc=1.454 g·cm-3,μ(Mo Kα)=1.784 mm-1,F(000)=1 248,R1=0.044 9,wR2=0.124 1(I>2σ(I)observed reflections)and R1=0.088 7,wR2=0.175 6(all reflections).A total of 4 885(Rint=0.032 1)unique reflections were collected,of which 2733 with I>2σ(I)were observed.The crystal structure shows that the central Sn atom is five-coordinated to assume a distorted trigonal bipyramidal configuration.The complex belongs to centrosymmetric dimer structure with four-membered central endo-cyclic Sn2O2units in which the bridging oxygen atoms are di-and tri-coordinated,respectively.In crystal,the molecules are packed in the unit cell in twodimensional network structure through the hydrogen bond(N…H-O)interaction between the O4-H atom and one of the nitrogen of an adjacent molecule.CCDC:715879.
organooxotin compound;synthesis;crystal structure;quantum chemistry
O614.43+2
A
1001-4861(2010)12-2160-05
2010-01-04。收修改稿日期:2010-04-20。
功能金属有机材料湖南省普通高等学校重点实验室(No.10K010)和湖南省高校“十一五”重点建设学科资助项目。
*通讯联系人。E-mail:hnkdz@yahoo.com.cn
邝代治,男,54岁,教授;研究方向:金属有机合成。
猜你喜欢
高中数理化(2022年16期)2022-09-14
椰城(2021年12期)2021-12-10
少儿科学周刊·少年版(2021年22期)2021-01-17
化工学报(2020年4期)2020-05-28
青岛大学学报(工程技术版)(2019年2期)2019-09-10
今日农业(2019年11期)2019-08-13
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
物理学报(2017年21期)2017-11-10
腐蚀与防护(2016年7期)2016-09-14
无机化学学报(2014年7期)2014-02-28
