
镍基催化剂:制备及水相催化糠醛加氢脱氧反应性能
2010-09-15 11:46:22张兴华王铁军马隆龙江婷刘琪英章
无机化学学报 2010年12期
张兴华王铁军马隆龙江 婷刘琪英章 青
(1中国科学院可再生能源与天然气水合物重点实验室,中国科学院广州能源研究所,广州 510640)
(2中国科学院研究生院,北京 100049)
镍基催化剂:制备及水相催化糠醛加氢脱氧反应性能
张兴华1,2王铁军*,1马隆龙1江 婷1,2刘琪英1章 青1
(1中国科学院可再生能源与天然气水合物重点实验室,中国科学院广州能源研究所,广州 510640)
(2中国科学院研究生院,北京 100049)
以SiO2-Al2O3、γ-Al2O3为载体采用浸渍法制备了不同负载量的镍基催化剂,以XRD、H2-TPR、NH3-TPD与低温N2吸附等技术对催化剂特性进行了表征,并进行了水相催化糠醛加氢脱氧实验研究以制备以C5为主要成分的生物汽油。重点考察了催化剂的镍负载量、载体种类及反应温度对糠醛转化率、目标产物C5选择性的影响,并对催化剂的重复利用性能和糠醛水相加氢脱氧反应机理进行了探讨。结果表明,Ni/SiO2-Al2O3催化性能明显优于Ni/γ-Al2O3。在140℃、氢冷压3.0 MPa的优化反应条件下,14wt%Ni/SiO2-Al2O3催化糠醛反应的转化率为63%,C5选择性高达93%。升高反应温度可以提高糠醛的转化率,但反应产物的选择性降低。催化剂在第3次重复使用时糠醛的转化率明显降低,催化剂表面有明显的残留有机聚合物和积碳,达到25wt%左右。
镍催化剂;加氢脱氧;糠醛;生物汽油
0 引 言
近年来随着化石资源的日益紧张,以及人类对能源资源需求的迅速增长,化石原料供给的稳定性变得日益脆弱[1-2]。因此,由可再生的生物质资源制取能源或化工品的探索研究已经成为当今关注的热点[3-4]。
呋喃类化合物如糠醛、甲基糠醛、羟甲基糠醛(HMF)等可以以较高的产率从木糖与果糖在固体酸、无机酸、离子液体等催化剂作用下脱水生成[5-7]。特别是糠醛,来源相当广泛,能从多种农业副产品比如玉米芯、锯屑、谷壳等中直接抽提[8-9]。目前,糠醛主要应用于糠醛树脂与精细化工品的生产。糠醛在合适的催化剂作用下可以选择性加氢生产糠醇、2-甲基呋喃或四氢-2-呋喃甲醛等[10-11]。由于糠醛含有醛基官能团,可以与丙酮通过羟醛缩合反应延长碳链,然后经过加氢和脱水反应得到液态的长链烷烃,该烷烃可直接用作车载燃料[12]。近年来Dumesic等[13-14]通过水相重整山梨醇制备以C5、C6为主的烷烃,直接高比例地加入到车用汽油中形成生物汽油,比传统的乙醇汽油能效更高。本工作探索通过加氢与脱水反应耦合将糠醛水相加氢脱氧直接转化为以C5烷烃为主(生物汽油的主要成分)的新工艺路径,这条新工艺路径提供了一条生物质经由呋喃类化合物平台生产烃类燃料及化学品的可能途径。不仅如此,纤维素乙醇工业中的生物质水解液中通常含有糠醛等毒物,不利于后续的发酵工艺[15],若利用水相加氢脱氧工艺将水解液中的糠醛转化为烷烃而自动分离,将为纤维素乙醇工业的发展解决一大技术难题。
镍是最常用的加氢催化剂之一,其相对廉价的制备成本、优异的加氢性能与较温和的反应条件使其在工业生产上应用极为广泛,在某些催化过程中,还显示出与贵金属相当的活性和选择性[16]。因此,我们在商业载体SiO2-Al2O3与γ-Al2O3上负载了金属镍,应用于糠醛水相加氢脱氧制备以C5为主的生物汽油的实验研究中。制备了一系列Ni/SiO2-Al2O3与Ni/γ-Al2O3催化剂,利用低温N2吸附法、XRD、H2-TPR等方法对催化剂进行表征,重点探索了催化剂载体、镍负载量、反应温度对糠醛转化率与产品选择性的影响,并对催化剂的重复利用性能进行了考察。
1 实验部分
1.1 催化剂制备
采用浸渍法制备负载型镍基催化剂。载体γ-Al2O3与SiO2-Al2O3为工业品,粒径小于75 μm,其中载体SiO2-Al2O3中SiO2含量为75%。制备过程如下:将一定量的Ni(NO3)2·6H2O溶于去离子水中,将一定量的载体粉末置于配制好的硝酸镍溶液中,超声波分散30 min,搅拌加热,直至水分蒸干,120℃下烘干4 h,马弗炉中于450℃焙烧5 h,粉碎。制备的催化剂分别以 Ni/γ-Al2O3和Ni/SiO2-Al2O3表示。催化剂使用前在450℃下用还原气(5%H2+95%N2)还原5 h。
1.2 实验装置与方法
在100 mL带机械搅拌的不锈钢高压反应釜中进行糠醛水相催化加氢脱氧反应试验。将预先还原好的催化剂2.0 g、去离子水40 mL、糠醛2.0 mL加人到反应釜中,通入氢气置换反应釜中的空气3次后,将压力升高至3.0 MPa。开启搅拌器,加热升温至预定温度时,开始计量反应时间与气体流量。气体反应产物以取样袋收集,并进行离线分析。
1.3 催化剂表征
NH3-TPD测试在自行组装的程序升温装置上进行,U石英玻璃管中装载100 mg催化剂,30 mL·min-1的He气流下以 10℃·min-1的速率升温至400℃并维持1 h,进行预处理。然后冷却至100℃进行NH3吸附,并在100℃下用He吹扫2 h,以除去物理吸附状态的NH3。以10℃·min-1的速率升温至 600 ℃;载气流量:30 mL·min-1。NH3的脱吸附量由TCD检测器监测。
H2-TPR测试在自行组装的程序升温装置上进行,U石英玻璃管中装载50 mg催化剂,30 mL·min-1的He气流下以 10℃·min-1的速率升温至400℃并维持1 h,进行预处理。冷却至100℃,以5%H2-95%N2(V/V)的还原气进行程序升温还原,以10℃·min-1的速率升温至800℃;还原气流量:30 mL·min-1。消耗的H2量由TCD检测器监测。
利用Micrometrics公司的ASAP-2010孔径分析仪对催化剂的比表面积和孔隙特性进行表征,N2吸附。利用德国耐驰公司的STA 409 PC Luxx的同步热分析仪对使用后的催化剂进行TG分析。分析条件:干燥空气,流速 30 mL·min-1;以 10 ℃·min-1的速率升温至850℃。
利用荷兰XPert Pro MPD X射线仪测定催化剂的物相结构。采用Cu Kα 射线(λ=0.15406 nm),工作电压(靶压)为 40 kV,工作电流(靶流)为 40 mA,扫描范围(2θ):10°~99°。该仪器采用 Peltier固体探测器,能量窗口范围 1.72~1.94 V,覆盖Cu Kα1波长范围。
1.4 产品分析与数据处理方法
以Shimadzu GC 2010型气相色谱仪(色谱柱为DB-5,30 m×0.25 mm×0.1 μm) 对气相产物进行分析。FID检测器温度200℃,毛细柱温度从50℃开始,2℃·min-1程序升温至70℃。外标法定量分析。
糠醛转化率定义为产物中的总碳物质的量与反应体系中所加入糠醛的总碳物质的量之比。糠醛转化率与产物分布(选择性)分别按如下两式计算:
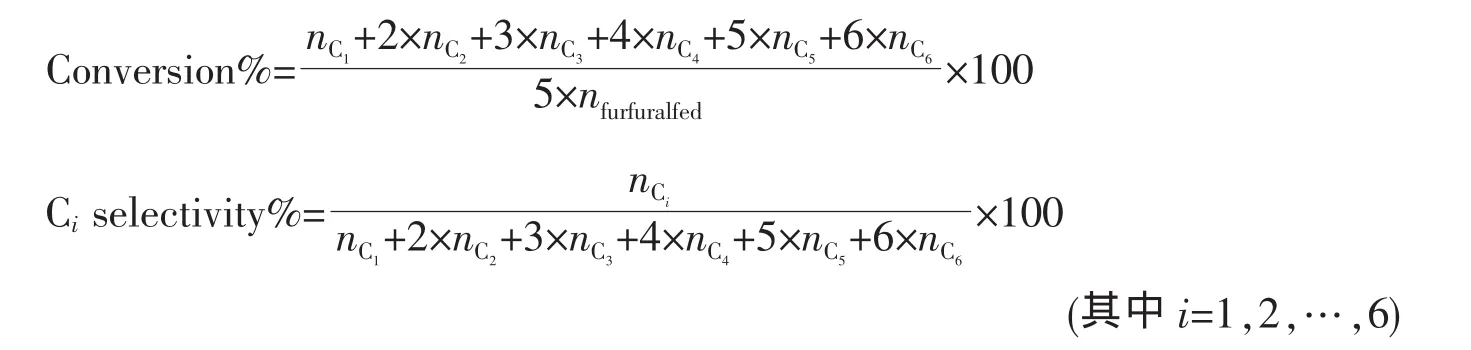
2 结果与讨论
2.1 催化剂表征
2.1.1 催化剂结构表征
表1为部分镍基催化剂的特征参数。由表1可以看出,随着Ni负载量的增大,催化剂的BET比表面积、孔体积与孔半径呈下降趋势。与Ni/SiO2-Al2O3催化剂相比,Ni/γ-Al2O3催化剂的BET比表面积、外部表面积、孔体积与孔半径数据明显要小。

表1 催化剂的孔特征结构Table 1 Porous structure characterization of the catalysts
2.1.2 XRD 表征
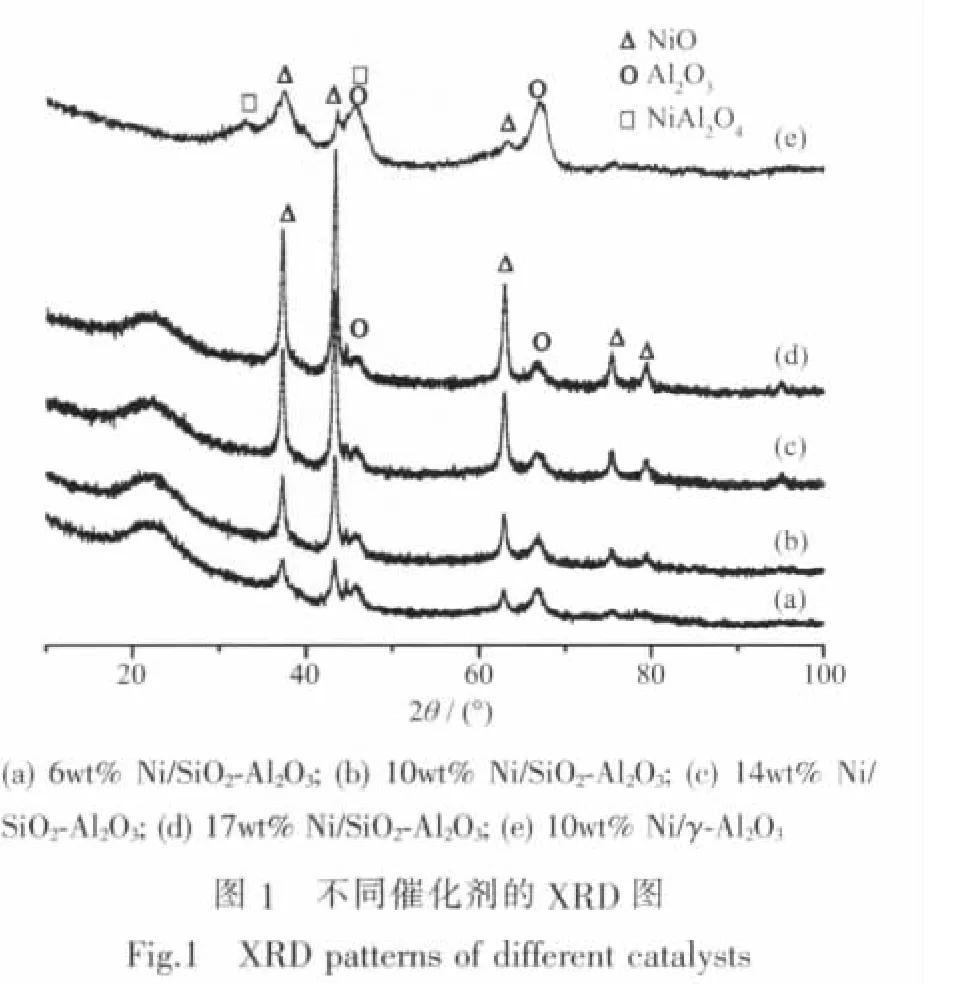
图1为不同催化剂样品的XRD图。从图中(a)~(d)可知,Ni/SiO2-Al2O3系列催化剂中,在 2θ为37.2°、43.2°、62.8°,75.3°、79.3°处出现了尖锐的信号峰,这些信号峰归属于NiO晶体的衍射峰,且随着Ni负载量的增大,衍射峰强度逐步增大(在Ni含量为 6wt%时,观察不到 75.4°、79.5°两处的衍射峰)。从(e)上可以看到,14wt%Ni/γ-Al2O3催化剂上NiO晶体在 2θ为 37.3°、46.4°、62.9°处的衍射峰相比 Ni/SiO2-Al2O3催化剂的衍射峰要弱很多,峰型也不尖锐; 而在 2θ为 75.4°、79.5°处的 NiO 晶体衍射峰则完全弥散消失。这说明NiO在γ-Al2O3载体上的结晶度较低。由于Ni/γ-Al2O3催化剂中的NiO与载体γ-Al2O3很容易发生作用生成 NiAl2O4,而 γ-Al2O3和NiAl2O4的XRD衍射线大多重合[17]。因此,仅通过XRD图尚无法推定NiO在γ-Al2O3表面是由于很好的分散作用而弥散,还是形成NiAl2O4尖晶石物相而被γ-Al2O3的XRD衍射峰掩盖。
2.1 3 H2-TPR 与 NH3-TPD 表征
图2为不同催化剂的TPR图。因为Ni2+还原为Ni0的过程中没有其他中间态,所以不同温度段的氢耗峰可以归属于不同状态的NiO还原产生的。对于Ni/SiO2-Al2O3系列催化剂,可以看到在370℃左右有1个小的低温还原峰,在450℃左右则有1个更大的还原峰。前1个峰可以归属为与载体间无相互作用的游离态NiO还原而产生的。在450℃左右观察到的还原峰,应该归属于分散态NiO还原产生的,因为NiO与载体间具有一定的相互作用使得分散态的还原能力下降,导致还原温度要高于游离态的NiO。一般而言,游离态的NiO经还原后生成晶粒大而比表面积小的Ni0晶粒,其催化活性较低;而分散态的NiO可以还原为小晶粒的Ni0,相对大晶粒的Ni0而言,增多了活性位,促进了催化剂活性的提高[17]。
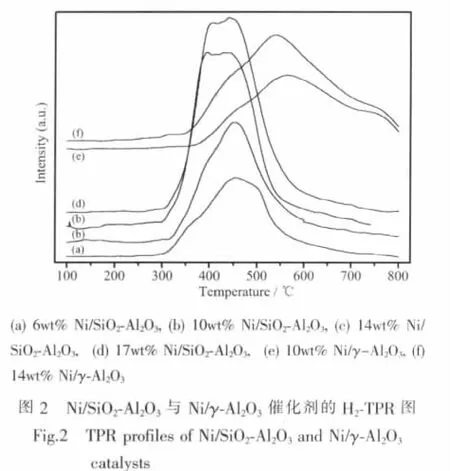
对于Ni/γ-Al2O3催化剂,除了450℃左右观察到的还原峰外,还在580℃左右观察到1个大的还原峰,根据文献[18]该峰可以归属为NiO与载体γ-Al2O3之间具有强相互作用,形成诸如NiAl2O4等形式存在的各类物质,这类型的物种需要较高的温度才能还原。结合XRD分析,可以证实NiO与载体γ-Al2O3之间形成了NiAl2O4尖晶石结构的物相。
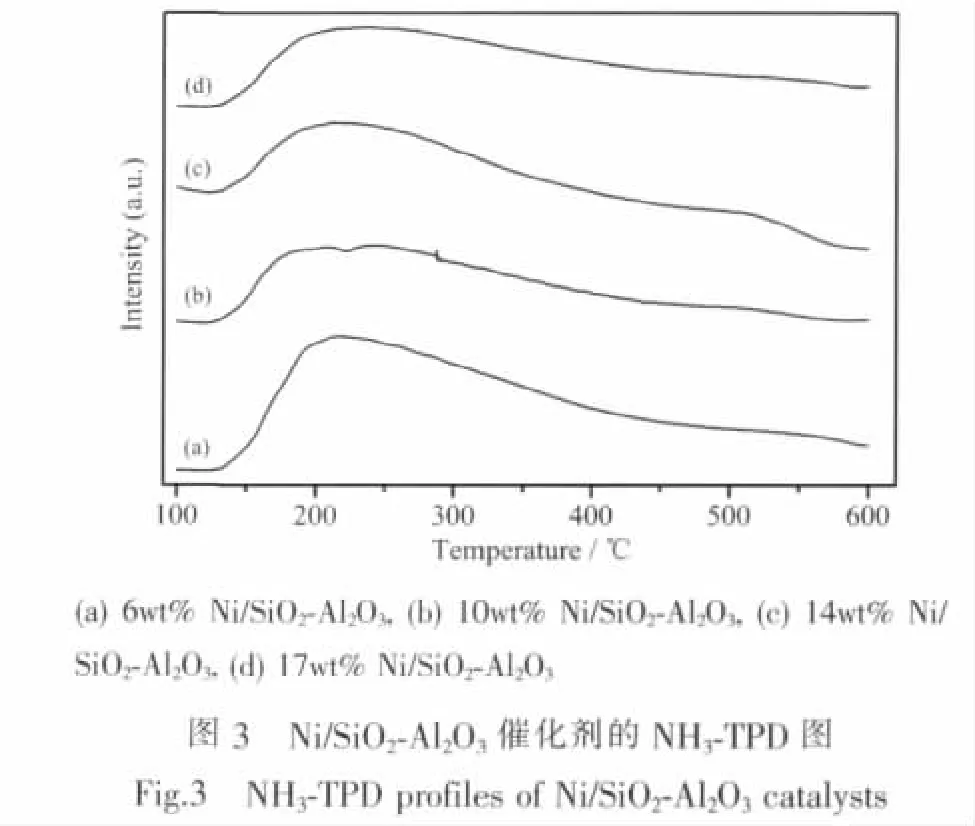
不同Ni负载量的Ni/SiO2-Al2O3催化剂的NH3-TPD见图3。从图可以看出,随着Ni负载量的增加,催化剂的NH3-TPD峰面积逐渐减少,即意味着催化剂表面的酸性位数量逐渐减少。据此可以推断Ni负载量增加,催化剂表面的酸性位会被逐渐覆盖。
2.2 水相催化反应性能
2.2.1 糠醛水相加氢脱氧的转化率
140℃反应温度下糠醛在不同催化剂作用下的转化率如表2所示。在相同的反应条件下,Ni/SiO2-Al2O3对糠醛的水相催化作用要明显优于 Ni/γ-Al2O3。其可能原因如下:一是如表1所示,催化剂Ni/SiO2-Al2O3的比表面积、孔径等均比γ-Al2O3大,有利于反应的进行与反应产物的扩散,促进糠醛转化反应的进行;二是H2-TPR研究揭示Ni/γ-Al2O3催化剂需要在更高的温度下才能较好的还原。而本工作中使用的催化剂均在450℃下还原,因此,未能充分还原也是导致Ni/γ-Al2O3催化剂活性较低的一个原因。为证实这一点,将10wt%Ni/γ-Al2O3催化剂在600℃下进行还原,然后在同样条件下催化糠醛加氢,发现糠醛转化率有大幅度的提升,增加至37.9%。但仍低于Ni/SiO2-Al2O3催化剂的活性。这表明未能充分还原是导致γ-Al2O3催化剂活性较低的一个因素。

表2 不同催化剂作用下的糠醛转化率Table 2 Conversion rate of furfural over different catalysts
从表2还可以看出,在Ni/SiO2-Al2O3的催化作用下,开始时,糠醛转化率随着催化剂镍负载量的增加而上升,在镍负载量为14wt%时转化率达到最大值63.0%;在镍负载量进一步增加到17wt%时,糠醛转化率反而下降至36.3%。Ni/γ-Al2O3催化剂也呈现出与此类似的变化规律。这可以解释为随着镍负载量的增加,催化剂上的活性组分随之增加,有利于糠醛转化率的提升;而当镍的负载量超过一定量后会发生催化剂载体的酸性位被覆盖,如NH3-TPD图所示,17wt%Ni/SiO2-Al2O3催化剂的酸性位数量明显减少,从而引起催化剂活性的下降。
2.2.2 糠醛水相加氢脱氧产物的选择性
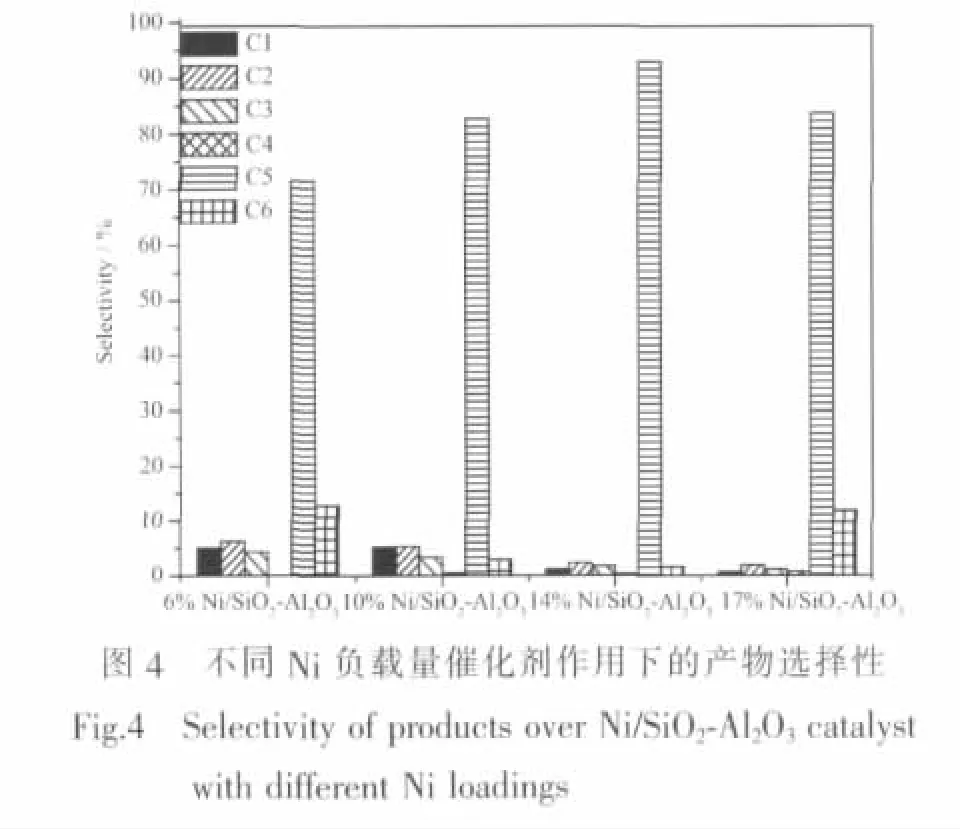
140℃的反应温度下,以负载量不同的Ni/SiO2-Al2O3催化剂分别催化糠醛进行水相加氢脱氧反应,其产物分布如图4所示。实验所用的催化剂对糠醛水相加氢脱氧反应均表现出良好的选择性。镍负载量对催化反应产物的选择性有一定的影响,在Ni/SiO2-Al2O3系列催化剂中,随着镍负载量的增加,产物中C5的选择性逐渐升高,在14wt%Ni/SiO2-Al2O3催化剂作用下,糠醛反应产物中C5选择性达到最高,为93%;镍负载量进一步增加到17wt%,反应产物中C5的选择性反而略有下降。这可以解释为镍负载量开始增大时,具有加氢催化作用的镍粒子增加,其与载体提供的酸性位协同作用促进C5产品的生成;但如NH3-TPD表征结果所示,当镍负载量增大到一定程度时,起脱水作用的酸性活性位数量就会减少,加氢反应与脱水反应二者不能很好的协同进行,从而导致产物选择性下降。
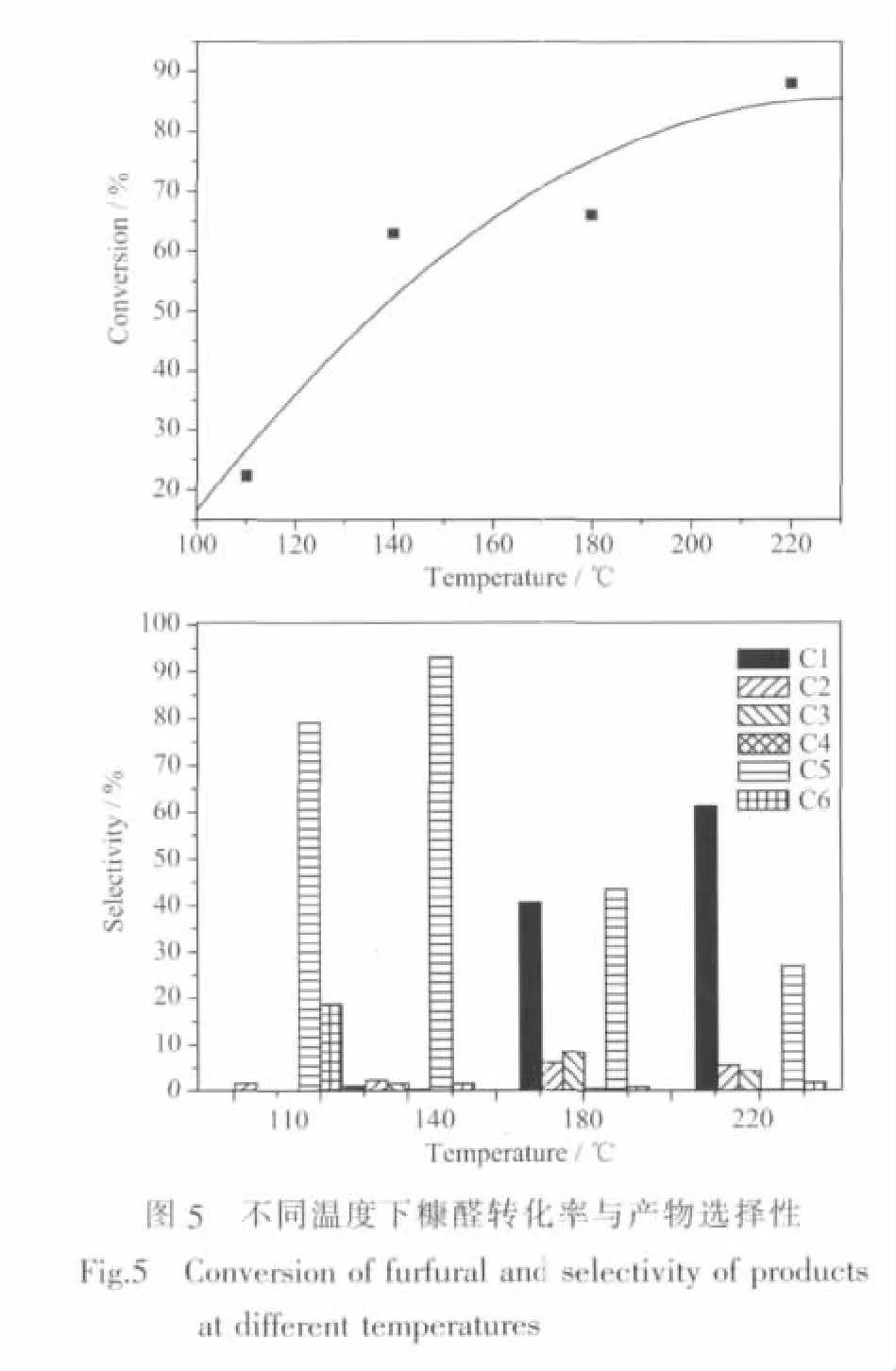
2.2.3 温度对糠醛水相催化的影响
反应温度是糠醛水相加氢脱氧反应的一个重要参数。优选的14wt%Ni/SiO2-Al2O3催化剂在不同反应温度下催化糠醛加氢脱氧反应的转化率与产物选择性如图5所示。在实验考察的范围内,随着反应温度的升高,糠醛的转化率明显提高。110℃时,糠醛转化率仅22.3%;当反应温度上升到220℃时,糠醛的转化率已经高达88.9%。但过高的温度,糠醛加氢裂解等副反应增多,反应产物的选择性变的不堪理想。如在反应温度为180、220℃时,反应产物中C1比例分别高达40.7%、61.3%,而目标反应产物C5所占比例仅43.5%、26.8%。由此可见,合适的反应温度,糠醛分子上官能团的活化、氢的解离过程与载体活性位的去羟基脱水过程相互耦合、协调进行,有利于促进糠醛向C5烷烃高效转化。
2.2.4 糠醛水相催化反应机理
在负载型镍基催化剂作用下,水相催化糠醛加氢脱氧制备烷烃的可能反应路径如图6所示。在金属Ni粒子作用下糠醛发生加氢反应,生成1-羟基四氢呋喃等反应中间物种,这些中间物种迁移至载体酸性位,发生去羟基脱水反应生成烯烃,再在金属Ni粒子作用下加氢生成烷烃并离开水相反应体系。

在产物中检测到了如 C1、C2、、C3和 C4等低碳烷烃。这些低碳烷烃是由糠醛加氢后的中间物种在催化剂作用下发生C-C键的断裂而形成的。低碳烷烃中的CH4也可能是反应过程中的CO和CO2在Ni作用下加氢生成的。此外,在产物中观察到了少量的C6烷烃,有文献[19]将类似的现象解释为反应过程中产生的CO、CO2与H2在含有酸性位的Ni基催化剂上发生费-托(F-T)反应(Fisher-Tropsch reaction)而生成的长链烷烃。
2.3 催化剂的重复利用性能
为了考察催化剂的可重复利用性能,优选的催化剂14wt%Ni/SiO2-Al2O3未经再生处理在140℃、3.0 MPa的冷氢压下重复使用。实验结果如表3所示,催化剂在第3次重复使用时糠醛的转化率出现明显的下降。这可以推测在前两次糠醛水相加氢脱氧反应过程中,催化剂已经逐步失活。糠醛在含有酸性位的催化剂存在下容易发生聚合反应形成聚合物,这些聚合物覆盖在催化剂表面是导致催化剂失活的可能原因之一。此外,在反应过程中形成的积碳覆盖了催化剂的活性位,也是导致催化剂失去活性的可能原因。

表3 14wt%Ni/SiO2-Al2O3催化剂的重复使用性能Table 3 Results for cycling of 14wt%Ni/SiO2-Al2O3catalyst
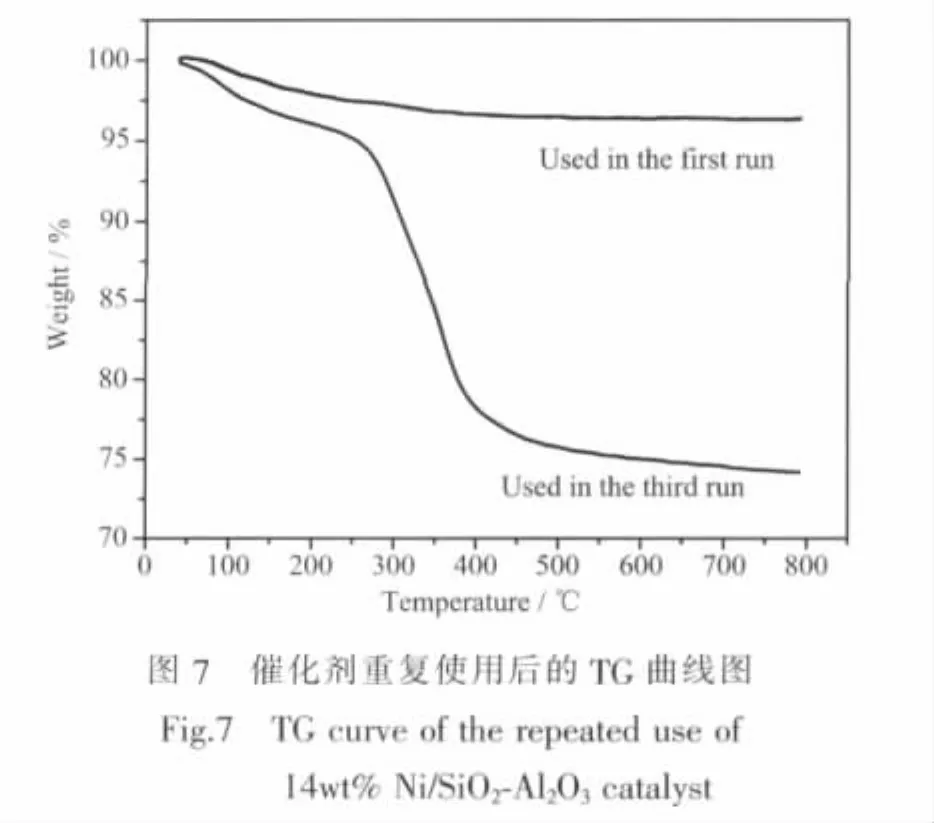
为了确认催化剂的失活原因,对使用过的两种催化剂进行热重分析表征,其TG曲线如图7所示。从图可以看到,使用1次后的催化剂失重曲线比较缓慢,总失重率也仅在4wt%左右。重复使用3次后的催化剂总失重率高达25wt%以上,在其TG曲线上可以看到,260℃之前的失重曲线比较平缓,失重率约为5wt%,可归属为催化剂上吸附的水、低沸点有机物等物质离开催化剂而产生的;而在260~390℃区间段失重曲线则相当陡峭,失重率约为18wt%,可归属为残留的聚合物等有机物燃烧而产生的;而390℃之后的平缓曲线则归属为催化剂表面积碳燃烧产生的。
3 结 论
催化剂载体和镍负载量对糠醛水相加氢脱氧反应的转化率与产物选择性有一定的影响。相对于Ni/γ-Al2O3催化剂,Ni/SiO2-Al2O3催化剂具有还原温度较低、比表面积与孔径较大的优势,因而更容易获得较高的转化率。在140℃,3.0 MPa H2压力下,14wt%Ni/SiO2-Al2O3催化剂催化糠醛反应的效果最佳,转化率达63.0%,C5的选择性达93%。提高反应温度有利于糠醛转化率的提高,但目标产物C5的选择性明显降低。适宜的反应温度为140℃。催化剂在第3次重复使用后的积碳与残留的有机物比较严重,达到25wt%左右,催化活性大大降低。延长催化剂的使用寿命及考察催化剂燃烧再生后的催化性能将是下一步工作的重点。
[1]Huber G W,Corma A.Angew.Chem.Int.Ed.,2007,46:7184-7201
[2]WANG Gang(王 刚),LI Wen(李 文),LI Bao-Qing(李保庆),et al.J.Fuel Chem.Technol.(Ranliao Huaxue Xuebao),2007,35(2):164-168
[3]Chheda J N,Huber G W,Dumesic J A.Angew.Chem.Int.Ed.,2007,46:7164-7183
[4]XU Ying(徐 莹),WANG Tie-Jun(王铁军),MA Long-Long(马隆龙),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,25(5):805-811
[5]Zhao H B,Holladay J E,Brown H,et al.Science,2007,316:1597-1600
[6]Binder J B,Raines R T.J.Am.Chem.Soc.,2009,131:1979-1985
[7]Román-Leshkov Y,Chheda J N,Dumesic J A.Science,2006,312:1933-1937
[8]Lima S,Pillinger M,Valente A A.Catal.Commun.,2008,9:2144-2148
[9]Sagehashi M,Nomura T,Shishido H,et al.Bioresour.Technol.,2007,98:2018-2026
[10]LIU Qi-Ying(刘琪英),LI Yong(李 勇),CAI Wei-Jie(蔡伟杰),et al.J.Mol.Catal.(China)(Fenzi Cuihua),2007,21(4):294-299
[11]WU Shi-Hua(吴世华),WEI Wei(魏 伟),LI Bao-Qing(李保庆),et al.Chinese J.Catal.(Cuihua Xuebao),2003,24:27-31
[12]Chheda J N,Dumesic J A.Catal.Today,2007,123:59-70
[13]Huber G W,Cortright R D,Dumesic J A.Angew.Chem.Int.Ed.,2004,43:1549-1551
[14]Huber G W,Chheda J N,Barrett C J,et al.Science,2005,308:1446-1450
[15]PAN Chun-Mei(潘春梅),SUN Xi-Yu(孙西玉),FAN Yao-Ting(樊耀亭).Liquor-making Science&Technology(Niangjiu Keji),2008,167(5):108-110
[16]Ji Y,Li W,Xu H,et al.Appl.Catal.A,2001,213(1):25-31
[17]ZHANG Yu-Hong(张玉红),XIONG Guo-Xing(熊国兴),SHENG Shi-Shan(盛世善),et al.Acta Physico-Chimica Sinica(Wuli Huaxue Xuebao),1999,15(8):735-741
[18]Ashok J,Raju G,Reddy P S,et al.J.Nat.Gas Chem.,2008,17:113-119
[19]Davda R R,Shabaker J W,Huber G W,et al.Appl.Catal.B,2005,56:171-186
Ni-Based Catalysts:Preparation and Catalytic Performance for Aqueous-Phase Hydrodeoxygenation of Furfural
ZHANG Xing-Hua1,2WANG Tie-Jun*,1MA Long-Long1JIANG Ting1LIU Qi-Ying1ZHANG Qing1
(1Key Laboratory of Renewable Energy and Gas Hydrate,Guangzhou Institute of Energy Conversion,Chinese Academy of Sciences,Guangzhou 510640)
(2Graduate School of Chinese Academy of Science,Beijing 100049)
Ni catalyst;hydrodeoxygenation;furfural;bio-gasoline
O614.83+3;TQ221.1
A
1001-4861(2010)12-2182-07
2010-06-07。收修改稿日期:2010-08-31。
国家自然科学基金(No.50776092);中科院知识创新工程重要方向性项目(No.KSCX2-YW-G-063)资助。
*通讯联系人。E-mail:wangtj@ms.giec.ac.cn
张兴华,男,30岁,硕士,助理研究员;研究方向:生物质液体燃料。
Abreact:The nickel-based catalysts for aqueous-phase hydrodeoxygenation of furfural were prepared by impregnation method using different supports and characterized by XRD,H2-TPR,NH3-TPD and low temperature N2adsorption The effects of support,nickel loading and reaction temperature on conversion of furfural and selectivity ofdesired C5products were studied.The reaction mechanism offurfuralaqueous-phase hydrodeoxygenation and the recyclability of catalysts were also discussed.The results showed that the activities of Ni/SiO2-Al2O3were obviously better than that of the Ni/γ-Al2O3under the same reaction conditions.The conversion of furfural over 14wt%Ni/SiO2-Al2O3catalyst was 63%and the selectivity for C5was as high as 93%under the optimization condition of 140℃and the cold H2pressure of 3.0 MPa.Conversion of furfural increased with increasing temperature,but the selectivity for desired C5product decreased with increasing temperature.The conversion of furfural decreased swiftly when the catalyst was re-used for the third time.About 25wt%residual cokes and polymers deposited on the used catalyst surface.
猜你喜欢
化学工程师(2023年1期)2023-02-17 15:09:48
石油沥青(2021年6期)2021-02-10 06:15:34
理化检验-化学分册(2020年12期)2021-01-26 00:41:38
中国果树(2020年2期)2020-07-25 02:14:28
石油地质与工程(2019年3期)2019-09-10 08:27:54
太原理工大学学报(2019年4期)2019-08-05 05:39:20
上海农业科技(2019年1期)2019-02-22 01:51:28
中国钼业(2019年2期)2019-01-19 15:54:06
水利技术监督(2016年6期)2017-01-15 14:01:33
化工进展(2015年6期)2015-11-13 00:27:04
