
不对称催化加氢的研究进展
2010-09-08 02:21焦广霞王维德赵
化工生产与技术 2010年6期
焦广霞 王维德赵 鹏
(华侨大学化工学院,福建 厦门 361021)
不对称催化加氢的研究进展
焦广霞 王维德⋆赵 鹏
(华侨大学化工学院,福建 厦门 361021)
叙述了不对称催化加氢的研究进展,分别对均相、多相不对称催化氢化以及均相催化剂的多相化进行了介绍,评述了均相、非均相不对称催化加氢的优缺点,提出了不对称催化加氢未来发展的方向和需要解决的问题,即一是如何得到更好的对映选择性,二是如何使催化剂具有更好的重复性。
手性;不对称催化;均相;非均相;加氢
手性是自然界本质特征之一,在生命科学和技术领域中手性物质作用重大。随着人们对自然和生命现象越来越多的理解,对手性化学品的需求逐渐增加,从而促进了合成手性化合物的研究与开发。通常手性化合物可以通过以下方法获得[1]:天然物提取,生物化学法,手性池法,消旋体拆分法,手性不对称催化法。由于前3种方法受自然条件和生物催化剂种类的限制,难以满足对手性化合物的巨大需求,而消旋体拆分法不经济,单一构型产物最大收率为50%,因此使手性不对称催化法成为获得手性化合物的重要方法。
不对称催化氢化是世界上第1个在工业上应用的不对称催化反应,由于其手型增值的突出优势而特别引人注目[2]。2001 年 Knowles、Noyori和 Sharpless因在不对称催化氢化研究中的杰出贡献而获得了当年的诺贝尔化学奖。不对称氢化加氢反应得到了迅速的发展,许多已经成熟地应用于工业生产,例如左旋多巴(L-Dopa)、萘普生(Naproxen)等的生产。具有光学活性仲醇是合成具有生物活性化合物的重要中间体,可以合成很多不同用途的有机化合物,如手性药物、农药和香精香料等[3]。通过前手性酮的不对称加氢反应是获得具有光学活性仲醇的重要途径。因此不对称催化加氢反应,无论在学术上还是工业应用方面都有很重要的作用。
不对称催化加氢反应又可以分为均相和非均相2类。
1 多相不对称催化氢化
1.1 传统过渡金属催化剂催化加氢
几乎所有的贵金属都可用作氢化反应催化剂,其中尤以镍、铂、钯、铑应用最为广泛。过渡金属的d电子轨道都未填满,它们表面易吸附反应物,有利于中间“活性化合物”的形成,且具有较高的催化活性,同时还具有耐高温、抗氧化、耐腐蚀等优良特性,在加氢反应中的应用相当广泛。大多数多相催化剂为载体负载贵金属及其合金,如Pt/Al2O3、Pd/C、Rh/SiO2、Pt-Pd/Al2O3、Pt-Pd/CaCO3、Pt-Rh-Al2O3等[4-5]。按载体的形状,负载型的贵金属催化剂又可分为微粒状、球状、柱状及蜂窝状。不同类型的催化剂有不同的制备方法,有浸渍法、共沉淀法、离子交换法、混合法和喷涂法等[6]。
纪红兵等人用氧化镍催化剂在液相中加氢还原羰基化合物,产率很高,且催化剂可反复使用9次而活性不变[7];赵会吉等人以负载型镍催化剂为加氢催化剂,液相加氢还原2-辛酮制备高纯度仲辛醇,2-辛酮转化率均可达到98%以上[8]。
贵金属催化剂以其优良的活性及稳定性而倍受重视,除在催化加氢应用外,还广泛用于脱氢、还原、异构化、芳构化、裂化、合成等反应。因此,其在化工、石油精制、石油化学、医药、环保及新能源等领域起着非常重要的作用。
1.2 由手性分子修饰的多相催化加氢体系
到目前为止,研究过的手性分子修饰的多相催化氢化体系很多,但比较成功的只有2个,一是酒石酸修饰的雷尼镍(Raney Ni)催化剂Ni-TA,二是生物碱修饰的负载铂催化剂体系。
1.2.1 TA-Ni体系
Izumi等在60年代初发现,将雷尼镍用光学纯的α-羟基酸或α-氨基酸处理后用于乙酸乙酯的加氢获得光学产物的光学选择性可达90%以上[9-10]:
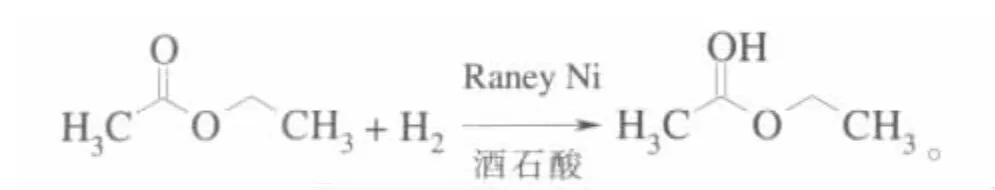
以R-构型的α-羟基酸或S-构型的α-氨基酸为修饰剂时,产物构型以R为主;反之,以S-构型的α-羟基酸或R-构型的α-氨基酸为修饰剂时,产物构型则以S为主。α-羟基酸的修饰效果比α-氨基酸好,而α-羟基酸中以酒石酸(TA)效果最佳。如果利用适量的羟酸,如羟基乙酸,在修饰前对雷尼镍进行预处理,则会使活性位的形成规整化,从而大大降低因制备条件变化对光学活性的影响[10-11]。在修饰液中加入适量钠盐,如NaBr、NaI和Na2SO4等,可使催化剂的光学选择性提高,其中以NaBr效果最佳[12-13]。
1.2.2 金鸡纳碱-铂体系
金鸡纳碱(Cinchona)是天然生物碱的一类,它们各由喹啉环单元和奎宁环单元所构成,2者之间由一个sp3杂化碳原子连接,其分子结构具有较大的柔韧性。
1978年,Orito报道成功地应用金鸡钠碱修饰负载型的铂催化剂以用于α-酮酯的不对称加氢[14]。目前,金鸡纳碱修饰的贵金属催化剂已广泛用于α-酮酯、酮肟以及一些不饱和酸类化合物中α-羰基、碳氮双键、碳氢双键的不对称氢化反应中。在这一催化剂上仍以α-酮酯的不对称加氢的效果最好,其中,丙酮酸甲酯或乙酯进行不对称加氢所得的光学选择性最高,对映体过量(ee)可达95%:

2 均相不对称催化加氢
早期的不对称催化加氢使用贵金属作催化剂,但ee很低。1965年G wilkinson发明了一种均相催化剂三苯基膦氯化铑(Rh(PPh3)3Cl),它能够很好地溶解于大部分有机溶剂在温和的条件下实现高活性催化加氢,与非均相催化剂相比有相当高的催化活性,这一发现为均相不对称氢化的发展提供了契机,从此手性配体加氢催化剂的合成成为不对称催化研究的前沿领域[15]。手性配体包括手性膦、手性胺、手性硫化合物等[16]。中心金属的选择多限于第Ⅷ族过渡金属,其中贵金属Ru、Rh和Ir应用最多。20世纪80年代以来。均相不对称催化加氢以其反应活性高、反应条件温和等优点在催化加氢领域得到广泛应用。
2.1 磷原子手性配体
磷原子和碳原子一样,具有空间四面体结构,可以形成手性化合物。含磷手性配体在不对称催化加氢反应中占有重要地位。手性磷配体又可以分为手性单齿磷配体和手性多齿磷配体[17]。
2.1.1 手性单磷配体
1968年Kownles和Horner各自独立使用含手性膦烷的铑催化剂实现了2-苯基丙酸和2-苯基丙烯的不对称加氢,尽管ee只有3%~15%,但是在使用金属配合物实现均相不对称催化氢化中起到开创作用[18-19]。这些单磷配体按成键种类可分为磷配体、亚膦酸酯配体、亚磷酸酯配体、亚磷酰胺酯配体以及其他类型的单磷配体。这方面已经有很好的综述[20]。这些配体不但易于合成、价格便宜、稳定性高,而且在许多不对称催化加氢反应中都有很高的选择性[21]。单磷配体能够以一配位、两配位甚至多配位的形式与中心金属离子配位并催化加氢反应。所以在某些反应中单磷配体比双磷配体更有优势。由于这些原因,单磷配体正在成为不对称催化加氢反应中一个新的研究热点。
2.2.2 等离子双极电切设备 常用4 mm 30°内窥镜,配合F24~F27电切镜鞘及电切环可用于多数膀胱肿瘤的切除。灌洗液使用生理盐水。相对于传统单极电切,等离子双极电切被认为可减少手术并发症风险(如由于闭孔神经反射所致的膀胱穿孔),并提供更好的组织标本,但结果尚有争议[4]。对于过度肥胖患者,有可能需要使用加长电切镜。
2.1.2 手性双磷配体
1971年,Kagan等合成了第1个手性双齿膦配体(R,R)-DIOP,其结构式为(Ph 为苯基)
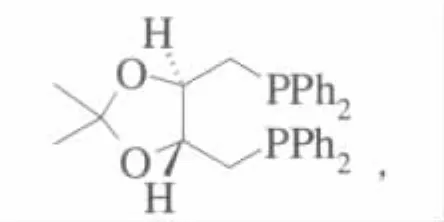
实现了手性膦配体设计的真正突破[22]。
早在上世纪70年代初,Kagan和Dang就已提出:如果一个具有C2对称轴的配体络合到中心金属离子上,则反应无论从哪一边靠近金属离子,它所接触的手性坏境都是一样的,换句话说,正是配体的对称性减少了反应的可能途径[23]。使用DIOP对脱氢氨基酸进行催化氢化,ee可以达到72%。此后各国化学家对DIOP进行了各种各样的改造,并成功运用于多种化合物的不对称催化加氢反应[24]。
1985年,Noyori成功地合成了著名的BINAP双膦配体[25]:
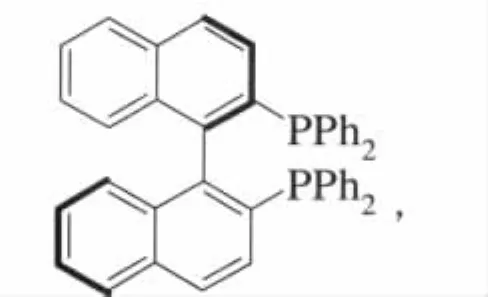
该分子并没有手性中心,而是依赖于手性轴使连结2个萘环的C—C单键旋转受阻,从而使整个分子具有旋光性,也使得双键间具有一定的灵活性。
与BINAP结构相似的联芳环类手性膦配体种类繁多,按手性轴键连的环来分有联萘、联苯、联杂环等几类手性膦配体,其中以BINAP衍生物为主的联萘类研究的较多。
Ishizaki等人将BINAP进行改造,所得配体的络合物可以在水中进行反应,从而使该体系的应用范围进一步扩大[26]。双齿配体形成的催化剂由于强的刚性,使得反应光学选择性提高,尤其是具有C2对称轴的。这种C2对称性可有效地减少过渡态的构象数量,使催化活性片段更加单一,因此在膦配体的开发方面,双齿配体倍受人们青睐。
2.2 其他配体
近年来,各国科学家设计出各种具有C2对称轴的氧原子手性配体,主要有噁唑硼烷系列、联苯系列、联萘系列、二茂铁衍生物、樟脑衍生物,糖类衍生物等[27]。同时也可使用手性配体对硼氢化试剂、铝试剂以及锌试剂等进行改造以实现不饱和官能团的不对称加氢。另外氮原子、硫原子手性配体在不对称氢化反应中的应用也得到快速发展[16]。
3 均相不对称加氢催化剂的多相化
研究表明均相催化加氢体系的选择性一般高于多相,活性高、条件温和。但是某些手性配体对氧气敏感、易分解,造成储存和重复使用的难度。多相不对称催化易分离回收,可多次使用,生产成本低[28]。而均相体系不易分离,而且分离过程中往往会出现络合物的破坏和配体的流失。随着催化反应越来越广泛的应用,人们为将均相催化剂的配体负载到非均相或可溶性的载体上进行了许多努力。早在上世纪30年代,Schwab和Stewart己经开始利用多相金属催化剂催化不对称合成反应,但由于多相催化往往受到载体性质的影响,光学选择性不好[29-30]。从上世纪70年代开始,人们将均相不对称催化剂与多相催化剂的优点相结合,实现了均相催化剂的多相化[31-33]。近年来,在均相手性催化剂的多相化在下列几个方面取得了一些进展:
1)在均相催化剂的基础上,通过不同的物理或化学吸附方法,将均相催化剂固定在无机载体上,如硅胶、沸石或硅氧烷薄膜,制备出固相化均相催化剂,希望它既保持均相催化剂高活性和高选择性的特点,又拥有多相催化剂易分离和易操作的优势[34]。
2)手性配体与高分子物质化学键合,先通过固相合成的方法将手性配体通过化学键合锚定到高分子树脂上[35];也可以先将配体做成单体,再发生自身聚合或者与其他高分子单体进行共聚,从而生成聚合的高分子手性配体,再与金属配位,形成化学键合在高分子上的手性催化剂[36]。
3)水溶性手性催化剂,研究较多的是水溶性磷配体,这类催化剂既能发挥均相催化剂的高活性、高选择性和反应条件温和等特点,又具有易与产物分离的两相特征[37-39]。例如,Davis等报道了固载水相催化剂,大大促进了在水溶性膦配体及两相催化体系中的研究[40-41]。随后,Davis对催化剂进行了改进,用乙二醇作为亲水多孔载体担载催化剂的液膜,有效地阻止配合物进入有机相中,大大提高了反应的收率和选择性。
一般而言,均相催化剂多相化技术可以保持均相催化剂的高反应活性、易重复性、反应的专属性、催化反应的可控性、催化剂的热稳定性等,更重要的是能简化反应后的催化剂与产物的分离、回收等操作。
4 展望
由于在分离、操作等方面的优越性,因此多相不对称催化氢化具有广阔的应用背景。但到目前为止,反应底物、手性修饰剂等的特殊性,使多相不对称催化氢化研究的体系仅局限于为数不多的几个。为了加速其工业化进程,迫切期待解决2个问题:一是如何得到更好的对映选择性,二是如何使催化剂具有更好的重复性。在未来的研究中以下几个方面值得重视:
1)研制手性催化新材料。例如选择具有规整表面或中孔的材料作为载体,将催化剂活性组分和手性修饰剂化学铆联到其内表面,也可以利用金属络合物的大小和特定的官能团,将金属络合物截留到具有层状材料的层之间,从而可以发挥分子筛孔道的立体择型效应和手性修饰剂表面的光学选择的双重功效。另外,由于树状高分子手性材料在反应体系中可以溶解,反应后可以通过改变溶剂、热沉淀、膜分离等简便方法分离,使其兼有高活性和高光学选择性的优点和容易分离的优点,其合成和应用近年来也逐渐引起人们的重视。
2)手性修饰剂的稳定。通过手性修饰剂(或手性配体)官能团和载体表面官能团之间形成的共价键,可以将手性修饰剂嫁接到催化剂表面;或者利用2者可以形成离子对作用,使手性修饰剂和载体相互作用,可能解决已有研究体系中手性修饰剂在催化剂表面吸附造成的容易脱落这一问题。运用分子烙印的方法,在载体表面形成具有一定光学选择性的手性环境,也是一个值得探索的方向。
3)理论计算和模拟。运用理论计算方法对反应体系中不同物种的构型、能量以及物种间的相互作用进行理论计算和模拟,对决定产物光学构型的过渡络合物的结构和能量进行理论计算,并结合多种谱学表征手段,探讨不对称加氢反应的机理和本质,从而更有效地设计新的多相不对称催化加氢反应体系。
4)拓宽催化氢化反应体系。选择合适的催化剂体系将反应类型拓宽到含C=C、C=N双键以及潜手性底物的不对称加氢反应等,可以进一步扩展研究领域。根据已有的研究经验设计和合成具有特定官能团和手性诱导中心的化合物,拓宽多相不对称催化领域中的手性修饰剂体系,也是一个值得研究的方向。
5)降低催化剂的污染。已报道的手性配合物大都用膦配体做协同配体,这类配体对空气敏感,环境污染也较严重。考虑以非膦配体代替膦配体做协同配体,合成对空气稳定无膦双官能团配合物非常必要。此类配合物对环境污染小,有着更为广泛的实用价值。
6)利用绿色化学过程。超临界流体或离子液体对反应物、产物具有较强的溶解能力,可以作为反应体系的介质,进一步提高体系的光学选择性,同时也满足绿色化学的要求。另外,生物催化加氢因其温和的反应条件和较高的立体选择性,在光学活性医药中间体的不对称合成中与传统化学合成相比具有很大优势。如果能比较方便地解决辅酶再生问题,有利于该类反应的产业化。
7)降低催化剂制作成本,提高重复使用效率。已报道的非均相催化剂和均相手性催化剂的中心金属研究主要集中在Ru、Ir、Rh等贵金属,花费较大,其应用有相当大的局限性。若利用廉价的金属(Mo、W、Ni和Fe等)作中心原子,将大大地降低工业生产中的成本。
[1]李月明,范青华,陈新滋.不对称有机反应[M].北京:化学工业出版社,2005:92-93.
[2]殷元骐,蒋耀忠.不对称催化反应进展[M].北京:科学出版社,2000:13-18.
[3]林国强,陈耀全,李月明.手性合成-不对称反应及其应用[M].北京:科学出版社,2007:420.
[4]E N Lipgart,J U Pesov,E I Klabunovskij.Asymmetric hydrogenation over Raney nickel modified with optically active substances.Effect of the reactionconditions on the degree of hydrogenation of acetoacetic ester[J].Kinet KataL,1971,12(6):1491-1494.
[5]A Hoek,W M H Sachtler.Enantioselectivity of nickel catalysts modified with tartaric acid or nickel tartrate complexes[J].J Catal,1979,58(2):276-286.
[6]Tokuma M Kitamura,R Noyori.Enantioselective synthesis of 4-substituted γ -lactones[J].Tetrahedron Let,1990,31 (38):5509-5512.
[7]Ji Hongbing,Huang Yueying,Qian Yu,et al.Ni-mediate liquid phase reduction of carbonyl compound in the presence of atmospheric hydrogen[J].Chinese J Chem Eng,2006,1(1):118-121.
[8]赵会吉,刘晨光,施敏.加氢还原法制备高纯度仲辛醇[J].石油大学学报,2002,26(3):94-103.
[9]IzumiY,ImaidaM,FukawaH,etal.AsymmetricHydrogenation withModifiedRaneyNickel.I.StudiesonModifiedHydrogenation Catalyst[J].Bull Chem Soc Japan,1963,36:21-25.
[10]Izumi Y.Modified Raney Nickel Catalyst:Heterogeneous Enantio-Differentiating (Asymmetric)Catalyst[J].Adv Catal,1983,32:215-273.
[11]Tai A,Harada T,Hiraki Y,et al.Stereochemical Investigation on Asymmetric Modified Raney Nickel Catalyst:Mode of Interaction between Modifying Reagent and Substrate in the Enantioface Differentiating Process[J].Bull Chem Soc Japan,1983,56:1414-1416.
[12]Harada T,Izumi Y.Improved Modified Raney Nickel Catalyst for Enantioface Differentiating (Asymmetric)Hydrogenation of Methyl Acetoacetate[J].Chem Lett,1978,18:1195-1199.
[13]Bostelaar L J,Sachtler W M H.The Role of Alkali Halides in the Enantioselective Hydrogenation of a Prochiral Keto Compound over Modified Nickel Catalysts[J].J Mol Catal,1984,27:387-391.
[14]Orito Y,Imai S,Niwa S,et al.Asymmetric Hydrogenation ofMethylPyruvate Using Pt-C Catalysts Modified withCinchonidine[J].Synth Org Chem Jpn,1979,37:173-174.
[15]Young J F,Osborn J A,Jardine F H,et al.Hydride intermediates in homogeneous hydrogenation reactions of olefins and acetylenes using rhodium catalysts[J].J Chem Soc,Chem Commun,1965,14:131-132.
[16]杨玉贵,吴帅.含硫手性配体催化的有机锌试剂在不对称加成反应中的最新研究进展[J].有机化学,2007,27(2):197-208.
[17]宓艾巧,孙建等,胡文浩,等.具有C2对称轴的手性配体及其不对称催化反应[J].合成化学,1995,36(1):19-28.
[18]KnowlesW S,SabackyM J.Catalyticasymmetric hydrogenationemploying a soluble,optically active,rhodium complex[J].J ChemSoc,Chem Commun,1968,17:1445-1446.
[19]Horner L,Siegel H,Büthe H.Asymmetric catalytic hydrogenationwith an optically active phosphine rhodium complex in homogeneoussolution[J].Angew Chem Int Ed Engl,1968,7:942-946.
[20]郭红超,丁奎岭,戴立信.不对称催化氢化的新进展——单齿磷配体的复兴[J].科学通报,2004,20(16):1575-1588.
[21]Lagasse F,Kagan H B.Chiral monophosphines as ligands forasymmetric organometallic catalysis[J].Chem Pharm Bull,2000,48:315-324.
[22]DangTP,KaganHB.Theasymmetricsynthesisofhydratropic acidandamino-acidsbyhomogeneouscatalytichydrogenation[J].J Chem Soc,Chem Commun,1971,20:481-486.
[23]Hawkins J M,Fu G C.Asymmetric Michael reactions of 3,5-dihydro-4H-dinaphth[2,1-c:l′,2′-c]azepine with methyl crotonate[J]J Org Chem,1986,51:2820-2825.
[24]Rettz M T,Kunisch F,Heitmann P.Chiral Lewis acids for enantioselective C—C bond formation[J].Tetrahedron Lett,1986,27(39):4721-4724.
[25]Noyori R,Ohta M,Hsiao Y,et al.Asymmetric synthesis of isoquinoline alkaloids by homogeneous catalysis[J].J Am Chem Soc,1986,108:7117-7119.
[26]Ishizaki T,Kumobayashi H.Water-soluble alkali metal sulfonate-substituted binaphthylphosphine transition metal complex and enantioselective hydrogenation method using it:EP,544455[P].1993-06-02.
[27]董建霞,杨定桥,刘二畅,等.手性双膦配体在不对称催化氢化反应中的最新进展[J].化工时刊,2005,19(3):41-45.
[28]左晓斌,刘汉范.多相不对称催化氢化的研究进展[J].分子催化,1997,11(4):309-320.
[29]Schwab G M,Rost F,Rudolph L.Optically asymmetric catalysis on quartz crystals[J].Kollooid-Zeitschrif,1934,68(2):157-162.
[30]Stewart T,Lipkin D.The asymmetric reduction of 13-methylcinnamic acid by β-glucose in the presence of Raney nickel[J].J Am Chem Soc,1939,61:3297-3300.
[31]Hetflejs J.Supported asymmetric hydrogenation catalysts[J].Smd Surf Sci Catal,1986,27:497-515.
[32]Blaser H U,Muller M.Enantioselective catalysis by chiral solids:approaches and results[J].Stud Surf Sci Catal,1979,59:73-92.
[33]Selke R,Capka M.Carbohydrate phosphinites as chiral ligands for asymmetric synthesis catalyzed by complexes[J].J Mol Catal,1990,63(3):319-334.
[34]丁奎岭,范青华.不对称催化的新概念与新方法[M].北京:化学工业出版社,2008:494-500.
[35]田卉,由宏君.不对称催化反应中固载催化剂的研究进展[J].精细石油化工进展,2005,6(4):25-32.
[36]Fan Q H,Ren C Y,Yeung C H,et al.Highly effective soluble polymer-supported catalysts for asymmetric hydrogenation[J].J Am Chem Soc,1999,121(32):7407-7408.
[37]Fan Q H,Deng G J,Chen X M,et al.A highly effective water-soluble polymer-supported catalyst for the twophase asymmetric hydrogenation:Preparation and use of a EG-bound-BINAP ligand[J].J Mol Catal A:Chem,2000,159(1):37-43.
[38]Fan Q H,Deng J,Lin C C,et al.Preparation and use of MeO-PEG-supported chiral diphosphine ligands:soluble polymer-supported catalysts for asymmetric hydrogenation[J].Tetrahedron:Asym,2001,12(8):1241-1247.
[39]Arhancet J P,Davis M E,Merola J S,et al.Hydroformylation by supported aqueous-phase catalysts:a new class of heterogeneouscatalyst[J].Nature (London),1989,339:454-455.
[40]Wan K T,Davis M E.Design and synthesis of a heterogeneous asymmetric catalysts[J].Nature (London),1994,370:449-450.
[41]Wan K T,Davis M.E Asymmetric synthesis of naproxen by supportedaqueous-phasecatalysts[J].JCatal,1994,148(1):1-8.
The Research Progress of Asymmetric Catalytic Hydrogenation
Jiao Guangxia,Wang Weide,Zhao Peng
(College of Chemical Engineering,Huaqiao University,Xiamen,Fujian 361021)
Asymmetric catalytic hydrogenation is one of the most important organic syntheses,which has developed rapidly in recent years.The paper summarized the research progress of asymmetric catalytic hydrogenation,homogeneous and heterogeneous-phase asymmetric catalytic hydrogenation was introduced,respectively.And how to hydrogenise homogeneous catalysts was also discussed.Through the comparison of merits and demerits between homogeneous and heterogeneous-phase asymmetric catalytic hydrogenation,the development direction and problems need to be solved were proposed here.
asymmetric;catalytic hydrogenation;homogeneous;heterogeneous-phase hydrogenation
TQ032.41
A DOI10.3969/j.issn.1006-6829.2010.06.010
福建省重点科研项目资助(2007Y0027)
* 通讯联系人,E-mail:wangwd@hqu.edu.cn
2010-10-16
猜你喜欢
分子催化(2022年1期)2022-11-02
第一财经(2019年8期)2019-08-26
材料科学与工程学报(2016年4期)2017-01-15
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
合成化学(2015年4期)2016-01-17
哈尔滨医药(2015年2期)2015-12-01
学习月刊(2015年14期)2015-07-09
物理化学学报(2015年5期)2015-02-28
郑州大学学报(理学版)(2014年3期)2014-03-01
