
气溶胶飞行时间质谱仪单颗粒质谱偏移问题及其纠正算法
2010-09-01 07:48:16王新宁
质谱学报 2010年3期
王新宁,陈 宏,杨 帆,杨 新
(复旦大学环境科学与工程系,上海 200433)
气溶胶飞行时间质谱仪单颗粒质谱偏移问题及其纠正算法
王新宁,陈 宏,杨 帆,杨 新
(复旦大学环境科学与工程系,上海 200433)
在单颗粒气溶胶质谱分析中,不同颗粒质谱图之间发生质谱偏移,谱峰质量数难以准确测定的现象已经被多次观察到。这种现象的存在使得应用单一的校准参数校准质谱图时,产生质谱峰辨认的不确定性。不同颗粒质谱图之间最大的偏移取决于电离激光光斑的大小。当校准参数不准确时,某质谱峰偏离正确m/z值的大小与该质谱峰的质量数呈线性相关。从理论上证明了这种偏移产生于颗粒物在激光光斑内电离位置的不同。为解决单一校准参数带来的不可避免的误差,提出了一种可编程的算法来自动找出每个颗粒物质谱的最优校准参数,以实现单颗粒质谱图的准确的质量校准。
气溶胶飞行时间质谱(ATOFMS);颗粒物质谱偏移;质谱校准;气溶胶
自从Prather课题组[1]1990年首次搭建气溶胶飞行时间质谱(aerosol time-of-flight mass spectrometer,A TOFMS)以来,ATOFMS已经在单颗粒气溶胶研究领域得到了广泛应用,并展现了很多传统气溶胶研究方法无法比拟的优势。传统的研究方法通常使用滤膜或撞击器来收集气溶胶样品,然后采用各种分析手段对其物理化学性质进行表征。从这种气溶胶的整体分析中无法得知单颗粒气溶胶的信息,也不能实现气溶胶的在线测定,这是传统气溶胶研究方法的弱点。A TOFMS从大气中直接采样,对单颗粒气溶胶进行瞬时分析,避免了传统方法的样品前处理或分析过程中不可预知的反应发生。ATOFMS能快速地测定单颗粒气溶胶,时间分辨率为秒量级,因此对各种大气事件,如风向的改变、局部污染事件的发生都可以反映出来。高的时间分辨率结合ATOFMS自身的可移动性,可以提供气溶胶的时、空分布信息。目前,ATOFMS已经被用来研究各种自然源气溶胶,如矿尘颗粒、海盐气溶胶、生物气溶胶等;人为源气溶胶,如黑炭颗粒、烟火燃烧颗粒、汽车尾气颗粒和各种实验室条件下产生的颗粒物等;以及环境大气气溶胶的演化研究等诸多方面[2-11]。
在针对中国东部城市大气气溶胶特性的研究中,利用ATOFMS开展了观测上海市区颗粒物的混和状态以及二次气溶胶形成机制等一系列工作[12-13],并对上海地区不同季节重金属颗粒物的主要来源与分布进行了分析[14-15],初步揭示出上海重污染条件下气溶胶的化学特征和特殊规律,显示出ATOFMS在气溶胶研究方面的重要价值。
质谱技术应用于单颗粒气溶胶的化学成分测定,主要是因为其对只有皮摩尔量级的单颗粒物质产生信号的能力[16]。一昼夜正常的大气采样将会有几万到几十万张的质谱图产生,如此庞大的数据量给分析工作提出了很大的挑战。大气气溶胶成分复杂,质谱出峰众多,为了避免质谱峰的误认,必须对每张质谱图准确校准,这是实现准确的质量校准的关键一步;但是,由于ATOFMS单颗粒质谱之间的相互偏移现象,对所有质谱图使用同一校准参数必然会产生误差。这里所说的质谱偏移是指当应用同一套校准参数时,不同颗粒物电离出的相同质量数离子在质谱图上显示不同的质量数。当这种现象发生时,颗粒物化学信息的分析误差是不可避免的。
这种偏移不但在ATOFMS上能观测到,而且在其他课题组搭建的类似的激光单颗粒质谱仪上都被观察到[17-18]。对使用的ATOFMS来说,如果能细心地筛选出一套校准参数,虽然有些质谱图发生了偏移,但大部分质谱图偏移并不太大,处于可接受的范围,只有相对较小的一部分颗粒物产生了足以产生质谱峰误认的偏移。消除这一部分误差的方法是对单颗粒质谱动态地选择最合适的质量校准参数,而不是应用同一套校准参数。因此,本工作提出一个解决此问题的途径,即自动地为每个颗粒物选择最合适的校准参数,该算法可通过编程实现,而免去手动校准的不便。在提出这个算法之前,将对质谱偏移的原因做出探讨和证明。
1 实验部分
1.1 仪器部分
A TOFMS(Model 3800,TSI Incorporated)进行气溶胶单颗粒分析的原理在文献[19-20]中有详细报导,在此仅进行简要描述。大气中的气溶胶颗粒经空气动力聚焦透镜进入仪器的真空部分,真空部分和外界气压之间的压力差促使气溶胶颗粒被逐渐加速,不同粒径的颗粒在空气动力作用下有不同的最终飞行速度。颗粒以此速度进入粒径测量区后,连续通过2个相隔一定距离的激光束而产生光散射,2个光电倍增管(PMT)通过测量2次散射光信号的时间延迟,可以确定颗粒的飞行速度。该飞行速度与颗粒动力学粒径有关,根据已知粒径的颗粒物所生成的速度-粒径校准方程,计算出气溶胶颗粒的空气动力学直径。随后,颗粒物进入飞行时间质谱区,颗粒的速度信息用来在精确的时间触发1个解吸/电离激光脉冲,使得颗粒物进入电离区时刚好被激光脉冲击中。颗粒在强激光作用下蒸发、电离,产生的正负离子分别进入反射式正、负离子飞行时间质谱仪,由微通道板(MCP)接收离子信号,高速数据采集系统同步记录颗粒的质谱和粒径数据。
1.2 质谱校准和数据处理
A TOFMS质谱的质量校准采用的是外标法,即通过已知化学成分的参考颗粒的质谱图去产生校准参数,然后将这些参数应用到其他质谱图的校准。ATOFMS附带的MS-Analysis软件也提供了手动输入校准参数的方法。参考颗粒物由已知组分的标准溶液通过气溶胶发生器(TSI Inc.,Model 9302 Atomizer)产生,标准溶液中含有Mo、Ag、Ba、K、Pb、Na、Li、V和HNO3等成分,总质量浓度900 mg·L-1。溶液谱峰可以直接判断出其对应的离子和质量数,这些离子的飞行时间分别被赋予相应的质量数。当输入6~7个质谱峰的质量数后,MS-Analysis会显示出一条质量校准曲线和一个形式为m=(at+ b)2的校准方程。此处,m为质量数(u),t为飞行时间(ns),a、b为校准参数。如果选择不同的参考颗粒物谱图,得出的 a、b值会有微小的差别,产生这种现象的原因是单颗粒质谱的偏移。正负质谱图分别单独校准,因此,a、b对正、负质谱图也是有差别的,这些参数被保存在校准文件中以便调用。需要说明的是,为了产生精确的质量校准,要求校准曲线的相关系数 R2大于0.999 99。
在MS-Analysis中所有的颗粒物质谱图都将被转化成质谱峰清单的形式。清单中的每一列对应该峰的峰面积、峰高、质量数、相对峰面积等,每一行对应一个质谱峰。MS-Analysis可以设定最低的信号强度以去掉噪声干扰。质谱峰清单被输入到 YAADA(http://www.yaada. org)数据库,以检索感兴趣的质谱图或颗粒信息。本工作用Matlab语言编写用于纠正产生偏移的 m/z数据程序,这个程序可以植入到YAADA程序中,方便地实施了重新校准,在质谱数据进入 YAADA数据库之前,为每个颗粒质谱图选取最合适的校准参数。
2 结果与讨论
2.1 质谱偏移的表现
典型的参考颗粒质谱示于图1。单颗粒的正负谱图都有几个稳定出现的质谱峰,虽然它们的绝对强度在不同质谱图中有很大的波动,但都较易辨认。在正谱图中稳定且易辨认的质谱峰有7Li+,23Na+,39K+,51V+,67VO+,138Ba+,208Pb+;负谱图中的有16O-,46NO2-,62NO3-,99Li(NO2)2-。为清楚地表明单颗粒质谱图之间的偏移现象,选取用单一校准参数校准之后的几张参考颗粒的质谱图,然后将它们叠加在一起,质谱图对比示于图2。图2中4种线形代表4个不同的参考颗粒物。每个小图上的4个质谱峰实质上是同一种离子产生的,但在应用同一套校准参数时,它们彼此分开,显示不同的质量数。
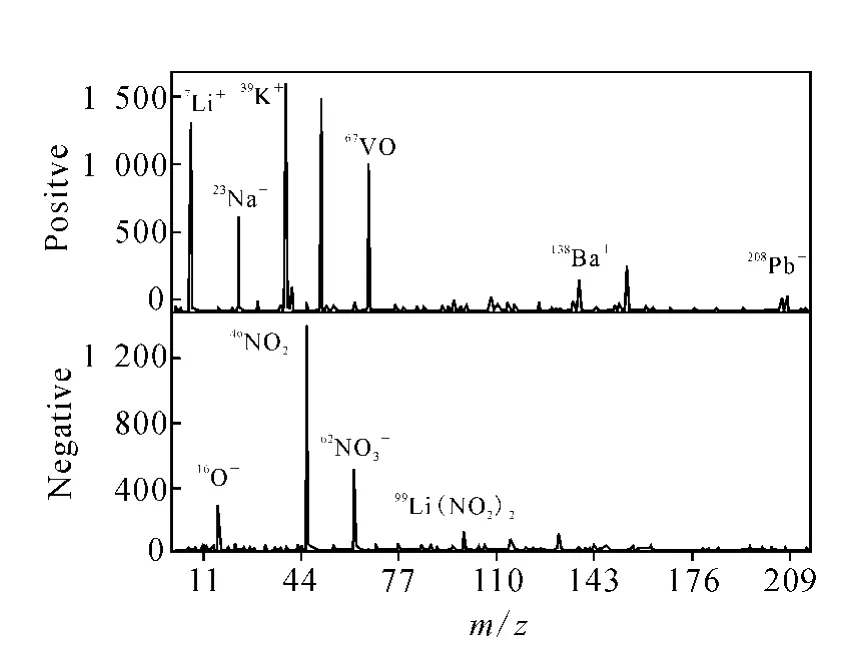
图1 典型的参考颗粒质谱Fig.1 Typical mass spectrum of a reference particle
图2中的11个质谱峰清楚地显示了质谱偏移发生的规律:不论离子的正负,质量越大的离子,其质谱峰分开的距离也越大,表明了质量大的离子有更大偏移的趋势。例如,7Li+离子峰4个颗粒物质谱中只有微小的偏差,而208Pb+离子峰在有些质谱图中显示的质量数却在206.5或209.6处。YAADA将这些非整质量数四舍五入到最近的整数。因此,为了不发生质谱误认,质谱偏移最大要控制在[m-0.5,m+0.5]区间内。但这种情况下,大质量的离子更容易产生校准错误,因为有些质谱的偏移已经超出了这个范围。误认的质量数将会进入 YAADA数据库,将不确定性引入后续的颗粒物归类和检索过程。
2.2 单颗粒质谱发生偏移的原因
Dienes等[21]已经推测颗粒物在激光光斑中电离位置的变动是导致质谱产生偏移的主要原因。相同的离子在同一套校准参数下显示不同的质量数,反映了这些离子在漂移管中有不同的飞行时间。将理论计算结果和机器实际记录的飞行时间数据比较,从理论上证明电离位置的微小变动对离子飞行时间的影响。图3可以直观地表示为什么颗粒物电离位置会产生偏移。颗粒物进入离子源时不是排列在一条线上,而是以一定直径的颗粒束式进入离子源。设颗粒束直径为D,同样,电离激光也具有一定的宽度,其光斑直径设为 d。在ATOFMS中,D的大小约为1 mm,光斑直径 d约为0.4 mm[22]。不同的颗粒物将在光斑中不同位置被电离,其最大的偏移是在激光光斑直径两端电离的颗粒,不同颗粒物在光斑中距离 S的最大值等于光斑直径d。图3中U1、U2、U3、U4分别为4个加速极板的电势,L为离子自由飞行的距离,l为内加速极板间的距离。不同电离位置使电离离子有不同的初始电场能,造成不同的飞行速度和飞行时间。
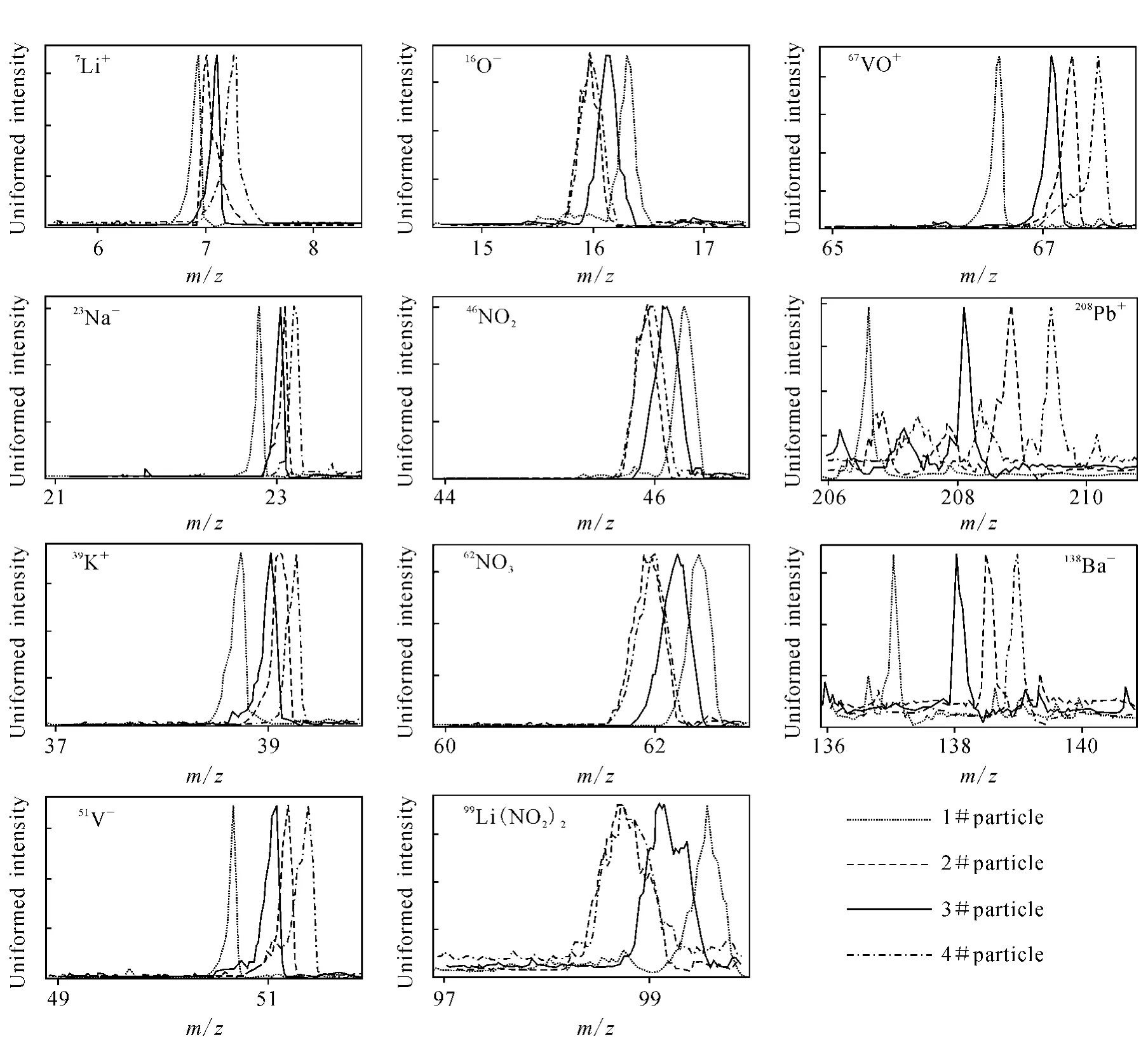
图2 采用单套参数进行质量标定后的4个颗粒的质谱图对比Fig.2 Comparison of mass spectra from four reference particles using single particle mass calibration
离子的飞行时间 T可以分为4个部分:在内加速极板间的飞行时间t1,在外加速极板和内加速极板间的飞行时间 t2,自由漂移的时间 t3,在反射极板间停留的时间t4。因此有:

为了考察2个不同初始位置(图3中圆形光斑中的2个黑点)离子飞行时间的差值,不需要计算每个离子总的飞行时间,只需计算出每个时间段的时间差,然后相加即可:

在这里采用2个近似来简化ΔT的计算。一个是离子初始速度的影响可以忽略不计,因为在2个位置电离的离子,其初始速度可以认为相同,因此设其初始速度均为0;另一个是Δt1和 Δt2本身对ΔT的贡献非常小,而且Δt1和Δt2在某种程度上互相抵消使它们的代数和更小,以至于可以认为贡献为零。对正离子来说,在左边位置电离的离子比右边的离子 t1值要小,而左边的离子因为有较小的电场能,离子在内加速极板和外加速极板间有更小的速度和更长的飞行时间,因此,如果Δt1为正,那么Δt2就为负,它们的和就更小,可以忽略。基于以上考虑,ΔT的计算可简化为:

假设2个离子的质量均为 m,电量均为 q,激光光斑(图3中圆形部分)在离子源中心位置。假设左侧离子的初始位置在光斑中心,而另1个离子向右偏移了 S距离(左侧离子是否位于光斑中心对ΔT的结果几乎无影响,该假设只是出于方程形式的简洁),它们最终的飞行速度分别为v1和 v2。在离子源中,离子的电场能转化为离子飞行的动能:
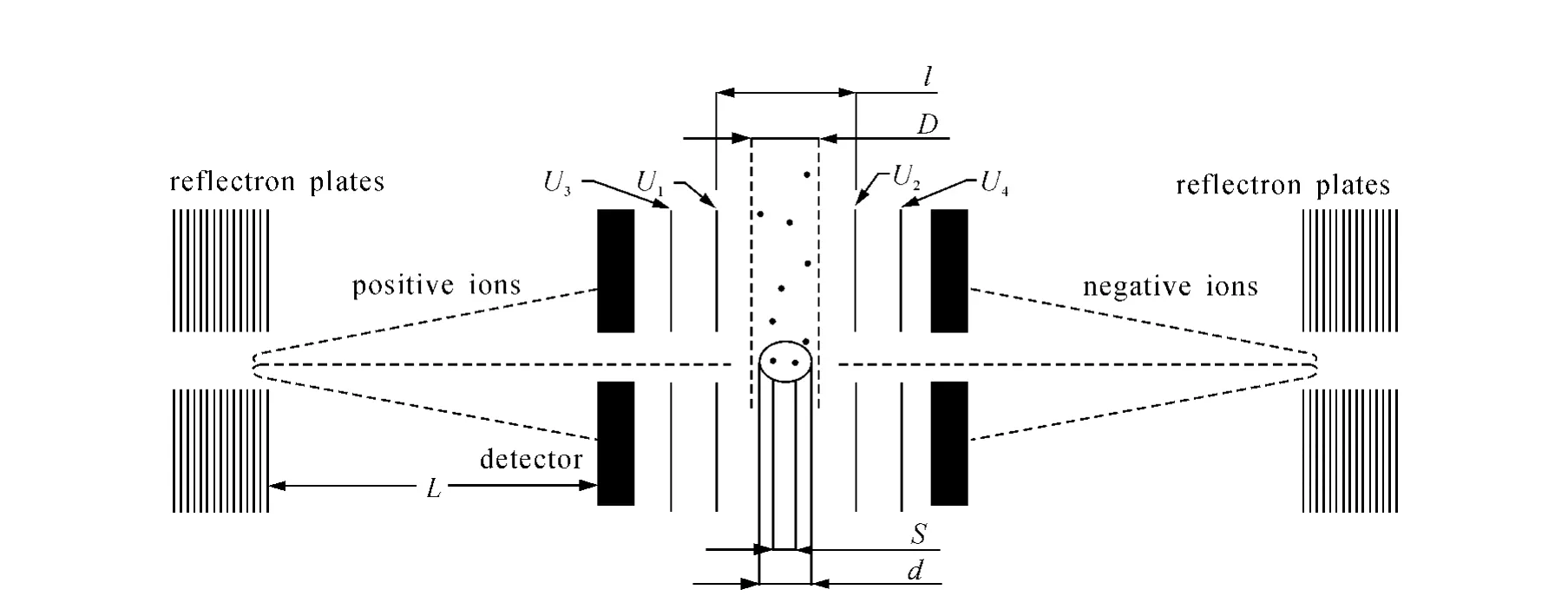
图3 ATOFMS飞行时间质谱部分原理示意图Fig.3 Schematics of the mass spectromety region of ATOFMS
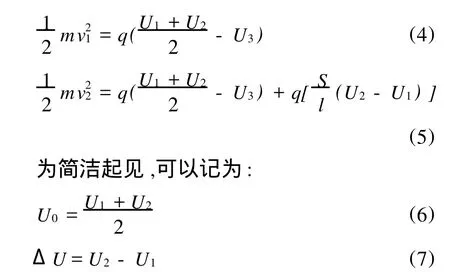
从以上式子可以解出2个离子的最终飞行速度:
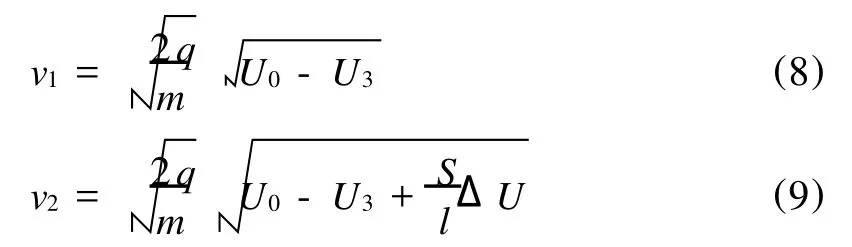
如果假设反射极板间的电场为 E,2个离子在自由飞行时间段的时间差Δt3和反射极板间的飞行时间差Δt4可以写做:

将式(8)、(9)代入式(10)、(11),考虑到ΔT=Δt3+Δt4,可以得到:
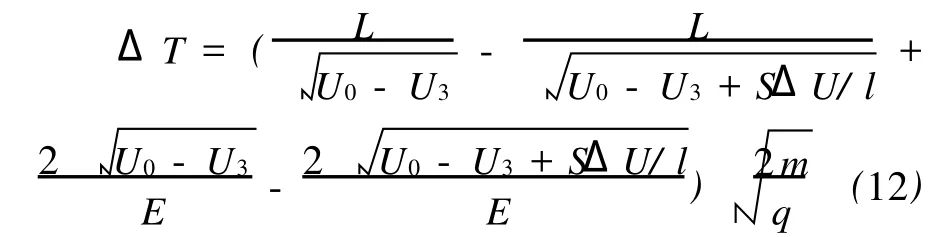
可以将式(12)的计算结果与实际质谱上的飞行时间偏移相比较。选择相距最远的电离位置即光斑直径的两端 (S取值为0.4 mm)作为偏差最大的位置,并代入ATOFMS其他相关的数值于式(12)中:L=0.606 m,l=0.01 m, U0=-814 V,U3=-6 078 V,ΔU=4 253 V, E=68 333 V·m-1。以钠离子为例,其质量m=23×1.66×10-27kg,电荷量 q=1.6× 10-19C,式(12)的计算结果为ΔT=68 ns,而实际质谱上钠离子飞行时间最大的差异为74 ns。同样,107Ag+离子根据式(12)的计算结果ΔT= 147 ns,实际观察值为162 ns。这其中的偏差可能是由于实际电离光斑直径略大于0.4 mm所致。从式(12)可以看出,在其他值固定的情况下,ΔT的大小由S唯一地决定,因此,离子飞行时间在不同质谱中的偏移与激光光斑的大小紧密相关。如果试图降低这种偏移,减小光斑直径是有效的方法,但这种方法势必会进一步减小颗粒的命中率,以损失更多的颗粒物质谱数据为代价。
质谱偏移中另一有趣的现象就是当质量校准参数不合适时,所有质量数的离子峰均将偏离实际的质量数,且这种偏移与离子质量呈直线关系。实际上,对特定的颗粒物来说,其电离位置固定,S也确定。式(12)中除了 m,其他所有的参数都是设定的常量,这相当于把其他所有常量合并成一个系数A:

ΔT造成的Δ m可通过对质量校准方程m=(at+b)2的微分得到:

式(14)表明,不正确的校准导致的质量偏移不但与离子质量呈线性关系,而且是正比例相关,这从图4可以得到验证。图4显示了使用固定校准参数时,部分颗粒物的质谱峰偏移与该质谱峰质量数的大小关系。图中6个颗粒的质谱分别代表了不同的偏移程度,且正负谱图都有同样的偏移趋势。虽然在低质量数的质量偏移很小,但这并不代表大质量数的离子也是这个情况。在有些颗粒物质谱上,处于100~200 u范围的离子其校准后的质量偏移已经超过1 u,这足以带来质量辨认的误差。如果一套校准参数来自某个参考颗粒的质谱,那么该参数严格地说只能适用于在同样位置电离的大气颗粒,它们之间的电离位置相距越远,在图4中将会表现出更大的斜率。

图4 使用固定校准参数时部分颗粒物质谱峰的偏移与该质谱峰质量数的大小关系Fig.4 Correlation between the mass deviation values and the actualm/zvalues was observed when spectrum is calibrated by constant factors
2.3 选择最优校准参数的算法
为了避免单颗粒质谱的校准偏差,必须改进所有颗粒质谱应用一套校准参数的方法,但是每一次实验将会有成千上万的质谱图,因此手动地校准每一张质谱图是不可行的。在文献[22]中曾提出寻找优化参数的办法:根据校准结果反算出该离子的加速电压,把加速电压乘以一个校正系数,然后用该电压得出优化的校准结果。校正系数的选择涉及质谱图曲线的积分问题,整数积分值最大者当选为理想的校正系数,文献[22]中没有具体说明如何操作,关于该方法只是一笔带过。该方法调节的电压值其实是参数 a,而不能调节b。参数 b与激光触发到离子产生之间的时间长短有关,对不同的颗粒物有不同的 b,因为蒸发电离所用的时间不尽相同,有机物质离子产生的时间因为包括蒸发这一阶段而较无机物质长。本工作提出一种不同的校准算法,可以为每张质谱图同时选择最优的 a、b。简单地说,如果考虑质量在m/z1~300范围内的所有离子质量数都处在整数附近,就能较容易地选择出最优的 a、b,步骤如下:
1)任意选择一个参考颗粒得出来的参数 a、b来校准某个实验颗粒质谱,从而获取该实验颗粒质谱图中所有质谱峰对应的飞行时间{ti}和质量数。
2)通过以下方法选择最优参数:分别为 a、b选择2个合适的变动区间和合适的增量Δa、Δb,让a、b分别以Δa、Δb为增量扫描整个变动区间,每一对 a、b都将飞行时间{ti}校准出一组质量数{mi},能产生最小 Y值的a、b就被选择为最优a、b。Y值的定义如下:

其中,〈mi〉为离mi最近整数。
3)用选择的 a、b重新校准质谱图。对特定的质谱图Y可以被认为是随校准参数a、b变化的二元函数。当 a、b在各自区间变化时,任一a、b组合对应的Y值也随着变化,当达到某一个a、b组合时,Y值达到整个区间的最低值,此时质谱图所有质谱峰偏离整数质量数最小。Y函数的图像大致形状示于图5。如果知道了其所有质谱峰对应的飞行时间,那么不同的校准参数就产生不同的Y值,而在所有可能的Y值中,产生最小Y值的那对校准参数才被认为是最准确的。图5是随机挑选的一个颗粒物的例子,这是一个变化复杂的曲面,在一个足够大的范围内, Y值总有一个最低点,这个最低点指示的就是最优的a、b组合。a、b的变化范围可以根据参考颗粒物产生的校准参数变动范围而定,并且一旦确定便无需更改。该范围反映的是实际上颗粒在激光光斑中电离位置可能的最大变动。
表1列出了一个任选颗粒物正谱图的校准结果,第1列表示质谱峰实际的质量数,第2列和第3列分别为用任意选定的恒定校准参数和本方法的校准结果。需要说明的是,负离子图谱也存在相似的偏移,并且正负离子的偏移方向是相反的。表1显示对于较大质量的离子,前面一种方法产生很大偏差,而本方法可以使质谱峰显示其实际离子质量数,而且校准结果非常靠近真实值。尽管这些数值不能被认为就是离子精确的质量数,但是在现有单颗粒气溶胶质谱分辨率的情况下,仍是一个很理想的近似结果。

图5 某特定实验颗粒质谱校准参数优化曲面图Fig.5 The optimization ofYas a binary function of calibration factor aandbThe lowest point indicates the best value

表1 固定校准参数和本文的校准方法在一个颗粒物质谱图上的校准结果对比Table 1 Comparison of the calibratedm/z values between the constant factor method and the newalgorithm present in this paper
3 结 论
本工作详细探讨了ATOFMS质谱偏移现象的起因,并提出了一种解决这个问题的理论算法。理论计算和实验结果说明,颗粒物电离位置的变动是颗粒质谱之间发生偏移的主要原因,并且质量数越大的离子发生偏移越严重。提出运用可编程算法来解决这个问题,即通过自动为每个颗粒质谱选择最优质量校准参数达到目的。本工作提出的算法简便易行,可以消除 ATOFMS质谱校准的不确定性,并且对同类型的单颗粒激光质谱仪的校准同样有效。
[1]PRATHER K A,NORDMEYER T,SAL T K. Real-time characterization ofindividual aerosol particles using time-of-flight mass spectrometry [J].Anal Chem,1994,66(9):1 403-1 407.
[2]SILVA P J,CARLIN R A,PRATHER K A. Single particle analysis ofsuspended soil dustfrom Southern California[J].Atoms Environ, 2000,34(11):1 811-1 820.
[3]BATES T S,QUINN P K,COFFMAN D J,et al.Marineboundary layerdustand pollutant transport associated with the passage of a frontal system over eastern Asia[J].Journal of Geophysical Research,2004,109:D19S23.
[4]NOBL E C A,PRATHER K A.Real-time single particle monitoring of a relative increase in marine aerosol concentration during winter rainstorms[J]. Geophysical Research Letters,1997,24(22): 2 753-2 756.
[5]FERGE T,KARG E,SCHRÖPPEL A,et al. Fast determination of the relative elemental and organic carbon content of aerosol samples by on-line single-particle aerosol time-of-flightmassspectrometry[J].Environ Sci Technol,2006,40: 3 327-3 335.
[6]GUAZZOTTI S A,SUESS D T,COFFEE K R, et al.Characterization of carbonaceous aerosols outflow from India and Arabia:Biomass/biofuel burning and fossil fuel combustion[J].Journal of Geophysical Research,2003,108:4 485.
[7]PHILIP J S,LIU D Y,CHRISTOPHERA N A, et al.Size and chemical characterization of individual particles resulting from biomass burning of local southern california species[J].Environ Sci Technol,1999,33(18):3 068-3 076.
[8]GROSS D S,GÄLLI M E,SILVA PJ,et al.Single particle characterization ofautomobile and diesel truck emissions in the caldecott tunnel[J]. Aerosol Science and Technology,2000,32(2): 152-163.
[9]LIU D Y,RUTHERFORD D,KINSEY M,et al. Real-time monitoring of pyrotechnically derived aerosol particles in the troposphere[J].Anal Chem,1997,69(10):1 808-1 814.
[10]GROSS D S,GÄLLI M E,SILVA P J,et al. Relative sensitivity factors for alkali metal and ammonium cations in single-particle aerosol timeof-flight mass spectra[J].Anal Chem,2000,72 (2):416-422.
[11]HU GHES L S,ALLEN J O,BHAVE P,et al. Evolution of atmospheric particles along trajectories crossing the los angeles basin[J].Environ Sci Technol,2000,34(15):3 058-3 068.
[12]WANG X F,ZHANG Y P,CHEN H,et al. Particulate nitrate formation in a highly polluted urban area:A case study by single-particle mass spectrometry in Shanghai[J].Environ Sci Technol,2009,43:3 061-3 066.
[13]YANG F,CHEN H,WANG X N,et al.Single particle mass spectrometry of oxalic acid in ambient aerosols in Shanghai:Mixing state and formation mechanism[J].Atmos Environ,2009,43: 3 876-3 882.
[14]ZHANG Y P,WANG X F,CHEN H,et al. Source apportionment of lead-containing aerosol particles in Shanghai using sinkge particle mass spectrometry[J]. Chemosphere,2009,74: 501-507.
[15]张雅萍,杨 帆,汪明明,等.运用单颗粒气溶胶质谱技术研究上海大气重金属(Zn,Cu)污染[J].复旦学报(待发表).
[16]NOBL E C A,NORDMEYER T,SAL T K,et al.Aerosol characterization using mass spectrometry[J].Trac-Tr ends Anal Chem,1994, 13(5):218-222.
[17]TAN P V,MALPICA O,EVANS G J,et al. Chemically-assigned classification of aerosol mass spectra[J].American Society for Mass Spectrometry,2002,13(7):826-838.
[18]MURPHY D M,MIDDLEBROOK A M,WARSHAWSKY M.Cluster analysis of data from the particle analysis by lasermass spectrometry (PALMS)instrument[J].Aerosol Science and Technology,2003,37:382-391.
[19]NORDMEYER T,PRATHER K A.Real-time measurement capabilities using aerosol time-offlight mass spectrometry[J].Anal Chem,1994, 66(20):3 540-3 542.
[20]GARD E,MAYER J E,MORRICAL B D,et al.Real-time analysis of individual atmospheric aerosol particles:Design and performance of a portable ATOFMS[J].Anal Chem,1997,69 (20):4 083-4 091.
[21]DIENES T.Development,characterization,and refinement of a transportable aerosol time-offlight mass spectrometer[D].California:University of California at Riverside,2002.
[22]TSI Inc.Model 3800 instruction manual,revision A[M].2004.
A New Algorithm to Correct the Particle-to-Particle Shift in Single-Particle Mass Spectrometry Analysis
WANG Xin-ning,CHEN Hong,YANG Fan,YANG Xin
(Department of Environmental Science and Engineering,Fudan University,Shanghai200433,China)
In single-particle mass spectrometry analysis,particle-to-particle shift was observed during many ambient aerosol studies.Chemical information uncertainties would be introduced into data set during mass calibration of spectra if constant factors were used. The shifting scale of particle spectra was closely related to the vaporization/ionization laser spot size under typical ATOFMS operation conditions.A linear correlation between the mass deviation values and the actualm/zvalues was observed when spectrum was calibrated improperly.This paper details this problem from a theoretical point of view and proposes a new algorithm to calibrate every single particle’s mass spectrum with the optimized parameters.
aerosol time-of-flight mass spectrometer(ATOFMS);particle mass spectrum shift;mass spectrum calibration;aerosol
O 657.63
A
1004-2997(2010)03-0179-08
2009-11-23;
2010-01-29
国家自然科学基金(40875074)资助
王新宁(1982~),男,硕士研究生,从事大气气溶胶研究。E-mail:wxn410@126.com
杨 新,教授,博士生导师。
猜你喜欢
都市(2023年6期)2023-12-28 07:56:54
诗歌月刊(2023年1期)2023-03-22 14:06:24
中学生数理化·自主招生(2022年10期)2022-05-30 10:48:04
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27 03:06:58
阅读(高年级)(2019年9期)2019-11-15 07:22:54
阅读与作文(小学高年级版)(2019年8期)2019-10-16 04:46:50
中学教学参考·理科版(2016年3期)2017-05-19 18:38:46
试题与研究·高考理综化学(2016年4期)2017-03-28 14:55:18
分析仪器(2016年1期)2016-03-04 11:17:00
分析仪器(2015年3期)2015-05-09 02:11:01
