
液相色谱-串联质谱法测定饲料中黄曲霉毒素的研究
2010-09-01 02:52朱聪英应永飞韦敏珏陈慧华陆春波周文海林仙军罗成江
质谱学报 2010年4期
朱聪英,应永飞,韦敏珏,陈慧华,陆春波,周文海,林仙军,罗成江
(浙江省畜产品质量安全检测中心,浙江杭州 310020)
液相色谱-串联质谱法测定饲料中黄曲霉毒素的研究
朱聪英,应永飞,韦敏珏,陈慧华,陆春波,周文海,林仙军,罗成江
(浙江省畜产品质量安全检测中心,浙江杭州 310020)
建立了液相色谱-串联质谱法测定饲料中6种黄曲霉毒素(包括黄曲霉毒素B1、B2、G1、G2、M1、M2)的方法。试样中的黄曲霉毒素经84%乙腈溶液超声提取后,用正己烷脱脂,过霉菌毒素多功能固相萃取柱净化,氮气吹至干,用流动相溶解后进行液相色谱-串联质谱法测定。方法的最低检出限为0.2μg·kg-1,定量限均为0.5μg·kg-1;标准工作液在0.5~100μg·L-1的范围内线性良好;加标浓度在1.0~100μg·kg-1范围内,饲料中黄曲霉毒素的回收率在66.1%~108%之间,批内相对标准偏差≤17.8%,能满足相关法规要求。
液相色谱-串联质谱法(LC-MS/MS);黄曲霉毒素;饲料
Key words:high performance liquid chromatography-tandem mass spectrometry(HPLCMS/MS);Aflatoxins;feeds
霉菌毒素(mycotoxins),又称真菌毒素,是某些霉菌在生长繁殖过程中产生的二次代谢有毒产物。在所有霉菌毒素中,黄曲霉毒素的毒性、致癌性、污染频率均居首位,是已知毒性最强的天然物质,如黄曲霉毒素B1敏感动物(鸭雏经口)的LD50=0.294 mg·kg-1,比氰化钾的毒性高10倍。黄曲霉毒素对饲料质量造成严重的影响,主要表现为致癌性、遗传毒性、致畸性,类激素中毒和白细胞缺乏症等,还会引起肾中毒、肝中毒、生殖异常以及抑制免疫反应,给畜牧业发展和畜产品安全造成极大危害,因此,我国明确规定了各种饲料中黄曲霉毒素B1的允许限量值[1]。
然而,要消除霉菌毒素造成的困扰却不容易,其中一个非常重要的原因是这些毒素的浓度通常很低,难以用常规方法测出。黄曲霉毒素包括B1、B2、G1、G2、M1、M2等多种形式。但是,目前有关这些毒素的检测方法很少,且以酶联免疫(ELISA)法和薄层色谱(TLC)法等筛选方法检测黄曲霉毒素B1为主[2-3],这些方法虽然在一定程度上解决了黄曲霉毒素的检测方法问题,但由于无法对样品进行准确的定性和定量分析,且容易出现假阳性等问题,不能作为最终的确证方法。
黄曲霉毒素的结构式示于图1。目前该类物质的检测方法主要包括免疫分析法[2],薄层色谱法[3]和液相色谱法[4-5]。其中免疫法往往作为筛选方法,不能作为定性依据;液相色谱法容易受到杂质干扰,且检测限很难达到要求;气相色谱-质谱法[6]作为定性方法,往往需要衍生化等步骤,操作较为繁琐;高效液相色谱-串联质谱(HPLC-MS/MS)是一种集高效分离和多组分定性、定量于一体的系统,已成为近年来研究黄曲霉毒素检测的主要方向,并已经应用到黄曲霉毒素的污染检测中[6-9]。本工作采用 HPLCMS/MS法测定饮料中的黄曲霉毒素,对样品前处理方法、液相色谱条件、质谱条件等进行系统优化,并对离子抑制效应问题进行初步研究。

图1 黄曲霉毒素的结构式Fig.1 Molecular structures of the Aflatoxins
1 实验部分
1.1 主要仪器
Waters Alliance 2695液相色谱仪-Micromass Quattro micro三重四极杆质谱仪,配有MassLynx V4.0软件;Sigma 3k18冷冻离心机; MS2minishaker漩涡混匀器;METTL ER XS-204电子天平;氮吹仪;Waters固相萃取装置等。
1.2 主要材料与试剂
黄曲霉毒素标准品购自 Fermentek公司,其中黄曲霉毒素B1、B2、G1、G2标准品含量≥99.0%,黄曲霉毒素M1,M2为10 mg·L-1的标准溶液,装量为1.0 mL。乙腈:色谱纯,购自美国Merck公司;实验用水为milli-Q超纯水;其他试剂均为分析纯;TC-M160多功能净化柱:购自美国 Trilogy公司。
1.3 标准溶液
1.3.1 标准溶液 分别精密称取5.0 mg黄曲霉毒素B1、B2、G1、G2对照品,置于100 mL棕色量瓶中,用甲醇溶解并稀释至刻度,即为 50 mg·L-1的标准品储备液,置于-18℃冰箱中保存。分别吸取0.20 mL上述标准品储备液和黄曲霉毒素M1、M2标准溶液各1支,置于同一10 mL棕色量瓶中,用甲醇溶解并稀释至刻度,即为1.0 mg·L-1的混合标准品工作液,冷藏保存。
1.3.2 基质标准曲线的制备 吸取一定量的上述混合标准品工作液,添加空白样品洗脱液,氮气吹干后,用V(0.1%甲酸溶液)∶V(乙腈)= 1∶1的溶液稀释成浓度为0.5、1.0、2.0、4.0、10.0、20.0、100μg·L-1的标准系列工作溶液。
1.4 仪器条件
1.4.1 色谱条件 色谱柱:Waters Atlantis dC18柱(3.0 mm×150 mm×3.0μm);柱温33℃;流动相:乙腈(A)和0.1%甲酸溶液(B);流速0.3 mL·min-1;进样量20μL。流动相洗脱梯度:0 min A 30%(φ),4.0 min A 45%(φ), 14.00 min A 100%(φ),15.00 min A 100% (φ),15.01 min A 30%(φ)。
1.4.2 质谱条件 ESI正离子电离源;毛细管电压3.0 kV;萃取电压2 V;离子源温度120℃;脱溶剂温度为380℃;脱溶剂气和锥孔气均为N2,其中脱溶剂气流速550 L·h-1,锥孔气流速50 L·h-1;碰撞气为高纯氩气,碰撞室的压力0.3 Pa。采用多反应监测(MRM)模式,母离子(Q1)/子离子(Q3)对均设为单位分辨,各离子对的驻留时间(dwell time)均为0.1 s,选择离子监测条件列于表1。

表1 MRM监测模式下黄曲霉毒素的监测条件Table 1 The optimized MRM conditions for Aflatoxins
1.5 样品处理
1.5.1 试样提取 称取(5±0.02)g试样于50 mL离心管中,准确加入25 mL 84%乙腈溶液进行提取,涡旋混匀2 min,置于超声波清洗器中超声提取20 min,中间振荡2~3次;取出,于8 000 r·min-1离心10 min,倾出上清液至分液漏斗中,加15 mL正己烷脱脂1次,待静止分层后,取下层溶液备用。
1.5.2 试样净化 取 TC-M160多功能净化柱,置于固相萃取装置上,准确量取5.00 mL下层溶液,控制流速2 mL·min-1左右,抽干,收集流出液,在 60 ℃下氮气吹干。用 1.0 mLV(0.1%甲酸溶液)∶V(乙腈)=1∶1的溶液溶解残渣,涡旋30 s,经0.22μm滤膜过滤后,上机测定。
2 结果与讨论
2.1 质谱定性定量分析
黄曲霉毒素属于脂溶性化合物,弱极性,以V(乙腈)∶V(水)=50∶50的溶液为基准流动相,采用“T”三通方式,对6种黄曲霉毒素的质谱条件进行优化,在正离子模式下进行全扫描,以选择合适的准分子离子峰和电离方式。黄曲霉毒素的准分子离子为正电离模式下获得的[M+H]+,结合基质空白和基质标准溶液的离子扫描图,优化锥孔电压、锥孔气、碰撞能量等仪器参数,将各监测离子的强度调谐至最大,从而相应地确定了各待测物在多反应监测模式下信号采集的特征离子对。霉菌毒素的子离子扫描图示于图2。
2.2 液相色谱条件
由于电喷雾质谱的电离是溶液状态电离,因此流动相的组成和配比不但影响目标化合物的色谱行为,还会影响到目标化合物的离子化效率,从而影响灵敏度。实验选择Waters Atlantis dC18柱(3.0 mm×150 mm×3.0μm)和Waters symmetry C18柱(2.1 mm×150 mm×3.5μm)为分离柱,在正离子模式下,流动相中加入0.1%甲酸溶液,有利于化合物的离子化。比较了2种色谱柱的分离效果,发现都能将被测药物与杂质较好的分离开,而 Waters Atlantis dC18柱比Waters symmetry C18柱效果更佳,因此选用前者作为分离柱。经过优化,确定了流动相比例和梯度条件,优化后的色谱条件见1.4.1。在此条件下,各组分的色谱行为良好,减少了样品的分析时间,达到了快速检测的目的。在该流动相条件下,被测物容易得到 H+而离子化,在很大程度上提高了检测的灵敏度。从加标样品的MRM色谱图(图3)可见,在待测物出峰的位置没有杂质干扰。
2.3 样品前处理条件
目前,霉菌毒素的提取方法主要以甲醇、乙腈以及它们与水配制成一定浓度的混合溶液作为提取剂。本实验在参考相关文献[7]的基础上,选择乙腈/水溶液作为提取液,比较了80%、84%、86%、90%乙腈/水溶液的提取效率,并对提取方法、提取时间等条件进行了研究。经过比较发现,采用84%乙腈/水溶液作提取液对各个霉菌毒素的总体提取效果最佳。
固相萃取净化方法是复杂基质中痕量检测进行净化时经常采用的方法,对不同净化柱的净化效果进行了比较。先后比较了Romer Multi-Sep 226型清洗柱/5mL、Romer MycoSep 226型清洗柱/5 mL和 Trilogy TC-M160多功能净化柱,根据各自的性能和厂家提供的方法优化了SPE净化条件。通过比较,3种多功能净化柱均能达到较好的净化效果,TC-M160更加稳定,因此,选择TC-M160作为饲料中黄曲霉毒素的净化柱。
2.4 线性方程和检测限
取1.3.2绘制标准曲线所用的系列溶液,按1.4建立的仪器条件进行测定。采用空白样品中添加目标化合物的方法,按1.5.1样品处理方法进行处理和检测,以 3倍信噪比为定量限(LOD),以10倍信噪比为检测限(LOQ),得到本方法霉菌毒素的LOD和LOQ。分别以选定的定量离子峰面积y对含量x(μg·L-1)做标准曲线,所得线性方程和线性相关系数列于表2。
2.5 离子抑制的影响
为了考察基质离子抑制对定量结果的影响,在1.0、5.0和50μg·kg-1的添加水平下,采用空白样品洗脱液加标和纯标准溶液进行比较,结果表明在进行痕量分析时,样品基质对待测药物的离子化影响较大,离子化效率在40%~79%之间。采用空白样品洗脱液添加标准品作为对照溶液,可以有效降低离子抑制对检测结果的影响,从而有效提高定量的准确性。
2.6 加标回收率和精密度试验
取豆粕、鱼粉、猪浓缩料、花生粕、鸡预混合饲料、鸡配合饲料作为代表性样品,添加适量混合标准工作液,使黄曲霉毒素的加标浓度在1.0~100μg·kg-1之间,按1.5的方法处理后,进行仪器分析。每个浓度点取5份样品,求其平均回收率,并计算批内相对标准偏差。

图2 霉菌毒素子离子扫描图Fig.2 Daughter ion scans of analytes
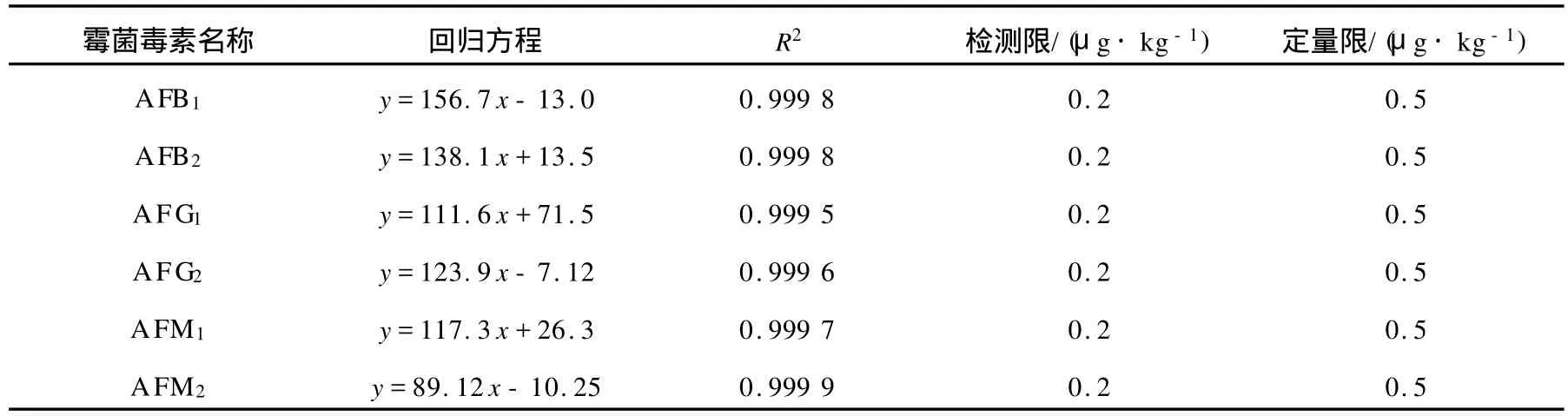
表2 黄曲霉毒素的线性方程、相关系数和检测限、定量限(n=6)Table 2 The calibration curves,corelation coefficient and LOD,LOQof Aflatoxins
实验结果表明,本方法建立的检测方法中,6种黄曲霉毒素的回收率在66.1%~108%之间,批内相对标准偏差≤17.8%。表3、4分别列出了豆粕、鸡配合饲料中霉菌毒素加标浓度为5.0~100μg·kg-1时的回收率结果。

图3 多反应监测模式下空白加标样品的离子色谱图(浓度20.0μg·L-1)Fig.3 Chromatograms of Aflatoxins by MRM mode in spiked samples(20.0μg·L-1)

表3 豆粕中霉菌毒素的添加回收率试验(n=5)Table 3 Result of recovery experiment in soybean meal(n=5)

表4 鸡配合饲料中霉菌毒素的添加回收率试验(n=5)Table 4 Result of recovery experiment in formula feed(n=5)
2.7 样品分析
应用建立的分析方法,对60份随机采集到的大豆粕、玉米蛋白饲料、鱼粉、玉米酒糟蛋白饲料(DDGS)和仔鸡配合饲料等样品进行测定,结果发现这些样品中普遍存在黄曲霉毒素污染,尤其以黄曲霉毒素B1的污染最为严重,检出率达到68.4%,浓度范围为2.5~45.2μg·kg-1,部分饲料的检测结果列于表5。该检测结果表明,本方法能满足我国对饲料中黄曲霉毒素污染进行监控的需要。

表5 部分实际样品中黄曲霉毒素的检测结果Table 5 Result of testing experiments in different feeds
3 结 论
本方法建立了液相色谱-串联质谱法同时测定饲料中6种黄曲霉毒素的方法,有效缩短了检测时间,扩展了检测毒素种类,节约了检测成本。在本实验的仪器条件下,被测样品中的各组分与样品基质均实现了较好分离,出峰时间合适,检测灵敏度较高。实验表明,该方法条件易于控制,结果准确、加标回收率稳定、重现性好,适用于饲料中6种黄曲霉毒素的定性、定量测定。
[1]全国饮料工业标准化技术委员会.GB/T 13078—2001饲料卫生指标[S].北京:中国标准出版社,2001.
[2]全国饮料工业标准化技术委员会.GB/T 8381—87饲料中黄曲霉毒素B1的测定方法[S].北京:中国标准出版社,1987.
[3]李秀芳,刘兴玠,胡 霞,等.酶联免疫吸附法(ELISA法)测定玉米中黄曲霉毒素B1的研究[J].卫生研究,1987,16(1):29-31.
[4]马 良,李培武,张 文.高效液相色谱法对农产品中黄曲霉毒素的测定研究[J].分析测试学报, 2007,26(6):774-778.
[5]PAPP E,HOTTA K,ZARAY G,et al.Liquid chromatographic determination of aflatoxins[J]. Micro-chemical J,2002,73(1/2):39-46.
[6]ZÖLL ER P,MAYER-HELM B.Trace mycotoxin analysis in complex biological and food matrices by liquid chromatography-atmospheric pressure ionisation mass spectrometry[J].J Chromatography A,2006,1 136(2):123-169.
[7]VENTURA M,GOMEZ A,ANAYA I,et al.Determination of aflatoxins B1,G1,B2 and G2 in medicinal herbs by liquid chromatography-tandem mass spectrometry[J].J Chromatog A,2004, 1 048:25-29.
[8]2002/657/EC Commission decision of 14 August 2002 implementing council directive 96/23/EC, concerning the performance of analytical methods and the interpretation of results,12.8.2002.
[9]The commission of the European Communities. Commission regulation(EC)No 472/2002 of 12 March 2002 amending Regulation(EC)No 466/ 2001 setting maximum levels for certain contaminants in foodstuffs L75/20.16.3.2002.
Determination of Aflatoxins in Feeds by Liquid Chromatography-Tandem Mass Spectrometry
ZHU Cong-ying,YING Yong-fei,WEI Min-jue,CHEN Hui-hua,LU Chun-bo, ZHOU Wen-hai,LIN Xian-jun,LUO Cheng-jiang
(A nimal Products Quality Testing Centre of Zhejiang Province,Hangzhou310020,China)
Totally 6 Aflatoxins in feeds were determined by high performance liquid chromatography combined with electrospray ionization triple quadrupole tandem mass spectrometry (HPLC-MS/MS)under the multiple reaction monitoring(MRM)mode,and especially focused on the optimization of extraction,clean-up,HPLC separation and MS/MS parameters of analytes.The Aflatoxins B1,B2,G1,G2,M1and M2were extracted by 84%of acetonitrile aqueous solution,purified by passing through multifunctional cartridges.The elution was evaporated by nitrogen blow and dissolved by mobile phase,and then assayed by HPLC-MS/MS.The limits of detection(LOD)are 0.2μg·kg-1,and the limits of quantitation(LOQ)are 0.5μg·kg-1.The calibration curves are linear between 0.5μg·L-1and 100μg·L-1for Aflatoxins B1,B2,G1,G2,M1and M2.Recoveries of those mycotoxins in feeds are 66.1%—108%,and inter-relative standard deviation(n=5)is less than 17.8%at spiked level of 1.0—100μg·kg-1.The method is simple,sensitive and reliable for feed safety.
O 657.63
A
1004-2997(2010)04-0240-07
2009-12-22;
2010-03-19
朱聪英(1963~),女(汉族),浙江武义人,高级畜牧师,从事饲料、畜产品安全检测方法的研究。E-mail:zjxm_zcy@163.com
应永飞(1977~),男(汉族),浙江仙居人,兽医师,从事兽药残留分析的研究。E-mail:yyf1001@163.com
猜你喜欢
煤化工(2022年3期)2022-07-08
当代水产(2022年1期)2022-04-26
湖南农业大学学报(自然科学版)(2021年4期)2021-08-13
现代畜牧科技(2021年4期)2021-07-21
色谱(2021年7期)2021-06-07
天然产物研究与开发(2018年9期)2018-10-08
山东工业技术(2016年10期)2016-09-06
中国资源综合利用(2016年10期)2016-01-22
中国钱币(2015年6期)2015-11-18
少儿科学周刊·少年版(2015年3期)2015-07-07
