
热加工食品中呋喃的研究进展
2010-08-27 11:12谢明勇黄军根聂少平
食品与生物技术学报 2010年1期
谢明勇, 黄军根, 聂少平
(食品科学与技术国家重点实验室,南昌大学,江西南昌 330047)
热加工食品中呋喃的研究进展
谢明勇, 黄军根, 聂少平
(食品科学与技术国家重点实验室,南昌大学,江西南昌 330047)
从当前备受关注的食品安全问题出发,较为全面地介绍了热加工食品中污染物呋喃的发现呋喃的毒理学。以及其形成途径和检测方法等方面的研究进展,并对上述研究进展进行了简要的分析和评述。
呋喃;热加工食品;形成途径;检测方法
呋喃(Furan)分子式为C4H4O,是一个具有芳香味与低沸点(31℃)的小分子环状烯醚,具有高度挥发性和亲脂性,容易通过生物膜并被肺或肠吸收,在人体中可引起肿瘤或癌变[1];呋喃还具有麻醉和弱刺激作用,吸入后可引起头痛、头晕、恶心、呕吐、血压下降、呼吸衰竭等症状,对肝、肾损害严重。国际癌症研究机构(International Agency for Research on Cancer,IARC)的研究结果表明,呋喃是鼠明显的致癌物,并将呋喃归类为可能使人类致癌物质的2B组[2];瑞典公共健康管理局和加拿大等的许多研究也发现了呋喃潜在的致癌危险[3-4]。2004年初,美国食品药品监督管理局(US Food and Drug Administration,FDA)的科学家意外地从一些食品中检测出了呋喃[5]。2004年5月,FDA发布:在很多经过加热处理的食品中检出了污染物呋喃;之后,欧盟食品安全局(European Food Safety Authority,EFSA)等也都报道从11大类的受检食品中发现呋喃[6-7]。鉴于食品中存在的呋喃可能会引起潜在的消费恐慌,2005年9月1日FDA出台了行动纲要,对食品中呋喃的暴露情况及其对人体的潜在影响进行深入研究。通过研究,FDA与EFSA得出一致结论,呋喃可能对人体致癌,为此启动了一系列的研究计划。此后,国外一些研究学者对热加工食品中呋喃的毒理学、前体物质、形成机理以及检测方法等方面进行了大量研究,并已取得一定的研究成果。我国在这方面的研究非常少,仅有极个别关于食品中呋喃含量的公开报道。为此,笔者在总结前人研究成果的基础上,结合最新研究动态,重点对热加工食品中呋喃的发现以及其毒理学等方面进行综述,并对其研究进展进行了简要的分析和评述,以期为我国相关领域对该污染物的进一步研究提供一些参考。
1 食品中呋喃的发现
与丙烯酰胺类似,由于呋喃广泛存在于许多类型的产品中(见表1),食品中的呋喃有可能成为一个严重的食品安全问题而引起潜在的消费恐慌[5-7]。
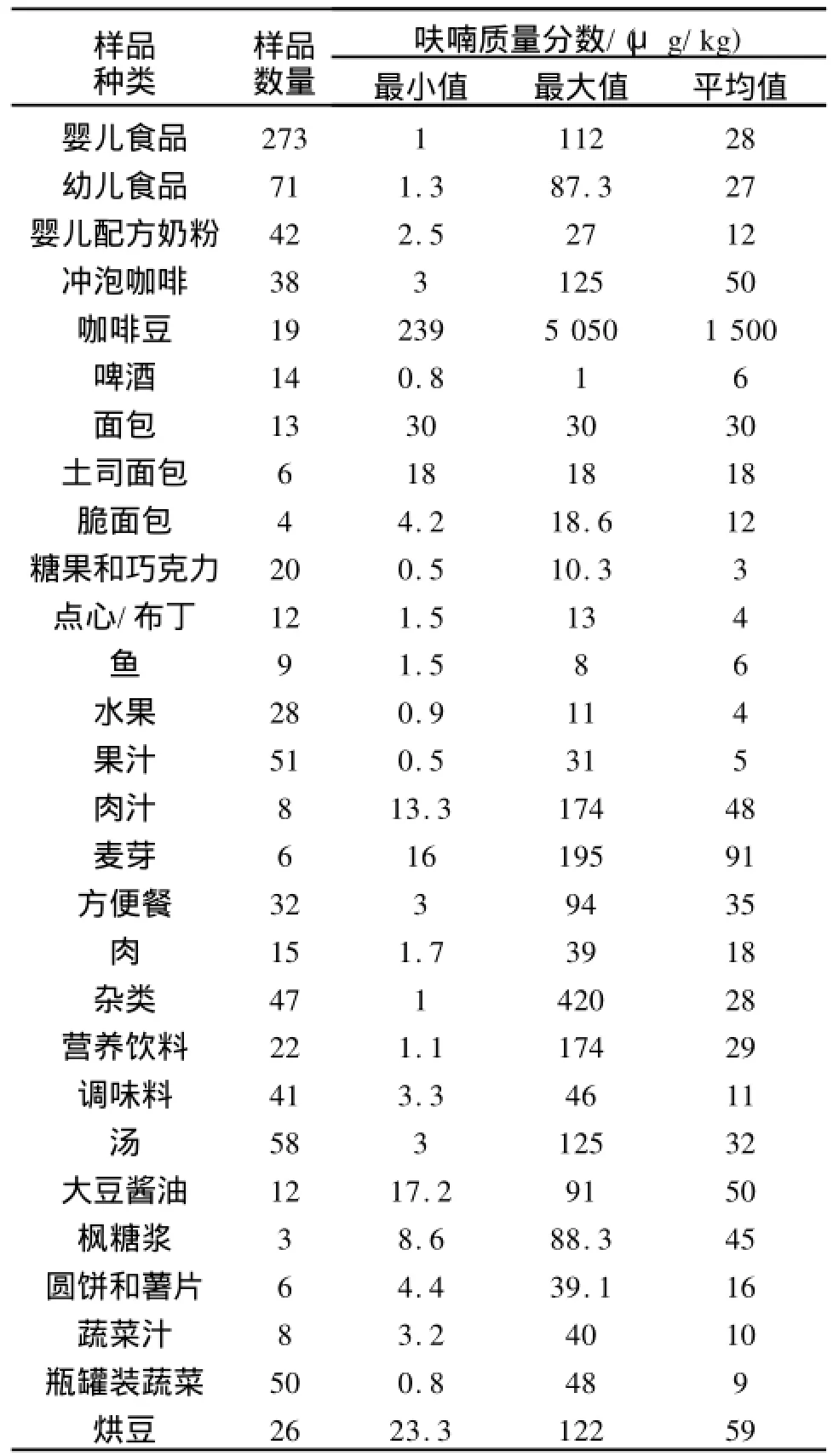
表1 EFSA和FDA报道的食品(多数是瓶装和罐装食品)中呋喃的含量Tab.1 Levels of furan in foods(mostly jarred and canned) reported by EFSA and FDA)
最早在食物中发现呋喃可追溯到1979年,Maga在咖啡中发现有呋喃存在[8]。在2004年初, FDA的科学家采用顶空进样-气相色谱-质谱联用技术从一些选取的罐装热加工食品中检测出了呋喃。2004年5月起,FDA[5]相继5次发布了食品中呋喃含量的数据:在很多经过加热处理的食品中检出了污染物呋喃,这些被检食品主要是婴幼儿食品、罐装蔬菜、豆类、水果、罐装肉和鱼、罐装酱、营养饮料和蜜饯、咖啡和啤酒等。继FDA之后,EFSA也开始调查食品中呋喃的暴露情况,在11个大类食品中都发现存在可检出的呋喃,经统计,呋喃质量分数超过100μg/kg的食品主要是咖啡、婴幼儿食品和调味料(如酱油)等3类食品[6-7]。其中,有96%的婴幼儿食品(273种食品中的262种)被检出呋喃,平均质量分数在28μg/kg。FDA研究人员发现,含有呋喃的食品几乎都是经过加热加工处理,其含量高的食品则大都是罐装食品。由于呋喃沸点只有31℃,所以研究人员试图通过食用之前加热这些食品以便让呋喃挥发,但是研究结果表明加热并没有明显地除去呋喃;此外,如果加热温度过高,食品中又将产生新的呋喃。我国食品中呋喃暴露情况等方面的研究罕见公开报道。
2 呋喃的毒理学
2004年,欧洲食品安全局(EFSA)公布了呋喃毒性的危险性评估,重要结论是呋喃对小鼠和大鼠具有明显的致癌性。之后,大量研究证实了这一结论[9]。美国国家毒理学计划(National Toxicology Program,N TP)[10]研究认为呋喃致癌的主要靶器官是肝脏,N TP的研究人员把呋喃溶于玉米油中进行动物试验,发现2年后造成大小鼠肝癌发病率增加,还可造成小鼠前胃鳞片状乳突瘤,肾髓质良性嗜铬细胞瘤[11]。目前对于呋喃的毒性研究还不完整,还没有呋喃致生殖和发育毒性的资料,也没有呋喃对人具体致癌机理的报道[12]。目前普遍的结论是呋喃可能对人体致癌。国际癌症研究机构(IRAC)已将呋喃归类为可能使人类致癌物质的2B组。
2.1 呋喃的代谢
呋喃能被迅速地吸入体内,并在体内高效排泄。放射性指示剂实验表明,在大鼠试验中,84%的单剂量口服呋喃在24 h内代谢完,其中有20%载于随尿液分泌的十多种化合物中,有22%随粪便排泄[13],剩下的被呼出。重复剂量的呋喃主要累积在肝脏和肾脏中,吸收的呋喃在细胞色素P-450 (CYP)酶的作用下迅速代谢,通过开环,形成二氧化碳和顺-2-丁烯-1,4-二醛[14],进一步与蛋白质和核苷结合[15],并且在放射性同位素示踪呋喃的鼠尿中发现其与单-谷胱甘肽共轭。含有人类肝细胞的初级培养物的实验表明,呋喃在细胞色素P450 2E1作用下的代谢非常之快。
2.2 呋喃的毒性
毒性研究表明呋喃是一个很强的致癌物质,影响到许多器官[10]。美国国家毒理学计划(N TP)的研究人员采用灌胃法将呋喃灌给大鼠和小鼠,服用剂量是20~160 mg/kg,16 d后发现两个物种的死亡率增加了,在13周后,低剂量呋喃造成大小鼠体重下降,其中,肝脏和肾脏质量增加,而胸腺质量减少,并导致大鼠和小鼠肝脏和肾脏的有毒病变,其严重程度与剂量成正比。大鼠有死亡,而小鼠没有死亡。研究也发现,持续较长时间将呋喃喂给小鼠,将导致小鼠体重的严重损失,并显著增加了肝细胞腺瘤和癌的发生;而给大鼠较高剂量的投药,持续13周每周5天以30 mg/kg呋喃灌胃,结果发现50只雄性大鼠出现了胆管癌,9个月后幸存的40只大鼠中的6只发现肝细胞癌,在15个月后幸存的10只雄性大鼠中均发现了胆管癌。
2.3 呋喃的遗传毒性
大多数研究学者认为呋喃的致癌性是由遗传毒性机制导致的。不管有没有S9(表面抗原-9)代谢活化,呋喃对某些鼠伤寒沙门氏菌株不具诱变性[10]。Lee和Bian等研究发现呋喃对TA100菌株有诱变[16]。也有文献报道,不管有没有S9活化,呋喃都对小鼠淋巴瘤细胞有诱变[17]。通过腹腔注射250 mg/kg的高剂量呋喃,将诱导小鼠骨髓细胞的结构染色体发生畸变,但不诱导姐妹染色单体交换;100~200 mg/kg的呋喃在活体内不诱导小鼠和大鼠肝细胞的DNA合成[10]。
顺-2-丁烯-1,4-二醛与α,β-不饱和化合物类似,会与DNA反应发生诱变性,它在无毒浓度下直接诱变对醛敏感的鼠伤寒沙门氏菌株(TA104),而不诱导其他几个菌株[18],这可能是因为呋喃或顺-2-丁烯-1,4-二醛能与靶细胞的DNA反应,并可以在呋喃诱导的肿瘤中发挥作用。
呋喃通过生物活化使ATP转化为代谢产物,从而激活了细胞毒性酶(包括核酸内切酶),这种酶会使DNA双链断裂,从而导致细胞死亡[19-20]。
然而,Louise和Kettil S[21]通过活体内和活体外的实验,得出了与上述结果不同的结论,即呋喃的致癌性是由非遗传毒性机制导致的。因此,呋喃的致癌机理还有待于进一步深入研究。
3 食品中呋喃的形成途径
已有文献资料表明,在热加工食品中,有多种途径可以形成呋喃,主要有:(1)还原糖单独存在时的热降解或与氨基酸共同存在时的美拉德反应[22]; (2)某些氨基酸的热降解反应;(3)抗坏血酸的热氧化作用;(4)多不饱和脂肪酸的热氧化;(5)类胡萝卜素的氧化[23]。图1总结了几种主要的前体物形成呋喃的途径[23-25]。
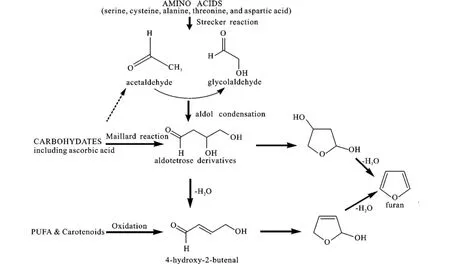
图1 几种主要的前体物形成呋喃的途径Fig.1 Proposed pathw ays and precursors of furan
Maga[8]研究发现食品中呋喃的主要来源是葡萄糖、乳糖、果糖等碳水化合物的热降解。Perez和Yaylayan[24]通过使用裂解GC-MS分析和13C标记建立糖、氨基酸和抗坏血酸模型系统进行了研究,发现氨基酸、糖、氨基酸/糖混合物和抗坏血酸等前体物质都可以形成污染物呋喃,其中抗坏血酸最容易产生这种物质。Becalski和Seaman[25]鉴定出了呋喃重要的前体物,如多不饱和脂肪酸,类胡萝卜素和抗坏血酸衍生物。据FDA报导,多种碳水化合物/氨基酸混合物或蛋白质模型系统(如丙氨酸,半胱氨酸,酪蛋白)和维生素(抗坏血酸、脱氢抗坏血酸、维生素B1)已被用来建造在食品中生成呋喃的研究模型[26]。
3.1 通过氨基酸的降解形成呋喃
Perez和Yaylayan[24]通过研究发现,有些氨基酸如丝氨酸(serine)和半胱氨酸(cysteine)不需要其他物质存在就可以通过热降解形成呋喃,它们都能代谢为乙醛(acetaldehyde)和羟乙醛(glycolaldehyde),然后通过醛醇缩合(aldol condensation)反应生成丁醛糖衍生物(aldotetrose derivatives),最终形成呋喃。然而,有些氨基酸,如丙氨酸(alanine)、苏氨酸(threonine)和天门冬氨酸(aspartic acid)不能单独形成呋喃,这些氨基酸仅能形成乙醛,并需要在还原糖、丝氨酸或半胱氨酸等存在下形成羟乙醛,然后通过以上过程形成呋喃。
3.2 通过碳水化合物的降解形成呋喃
在缺乏氨基酸的情况下对糖进行加热时,呋喃主要是由完整的糖骨架形成,甲酸和乙酸被确定为糖降解过程中的副产品,这就说明了己糖在C1和/或C2处发生了裂解。然而,丙氨酸、苏氨酸、丝氨酸的存在,可通过C2片段(如乙醛和羟乙醛)的重组促进呋喃的形成,这些C2片段可能源于糖和氨基酸。在水溶液中,大约一半的呋喃是由糖片段的重组产生的[27]。
Perez和Yaylayan[24]通过相应的研究发现,碳水化合物可通过4种途径(A,B,C,D)降解为丁醛糖衍生物,而后,丁醛糖衍生物通过环化作用形成呋喃。在氨基酸存在下,还原性己糖(Hexose)发生美拉德反应,形成活性中介物质1-脱氧邻酮醛糖(1-deoxy-osone)和3-脱氧邻酮醛糖(3-deoxy-osone) (如图2,途径A、D):1-脱氧邻酮醛糖必须通过α-二羰基键断裂形成丁醛糖(aldotetrose);3-脱氧邻酮醛糖经过α-二羰基键断裂,接着氧化和脱羧生成2-脱氧丁醛糖(2-deoxy-aldotetrose);而己糖在没有氨基酸存在时,可通过裂解过程形成丁醛糖(如图2所示的途径B),只是含量少。途径C表明己糖通过脱水反应和反醛醇裂解过程可形成2-脱氧-3-酮基丁醛糖(2-deoxy-3-keto-aldotetrose)。如图2所示,以上所有的丁醛糖衍生物很容易通过环化和脱水作用形成呋喃。
3.3 通过抗坏血酸形成呋喃
通过建立模型系统(在118℃条件下加热30 min)的实验表明,抗坏血酸(ascorbic acid)衍生物也可以生成呋喃。然而,脱氢抗坏血酸和异抗坏血酸生成的呋喃是抗坏血酸的10倍。一般情况下,含有钠盐的酸较其游离酸产生更少量的呋喃。增加三氯化铁不会影响游离酸生成呋喃的量,但会显著增加其相应的钠盐形成呋喃的含量。虽有文献指出,抗坏血酸在180℃条件下加热可以生成乙醛和乙醇醛,在理论上,也就可以通过羟醛缩合产生呋喃,但是,由于食品中抗坏血酸易于氧化和水解随后形成2,3-二酮古洛糖酸(DKG)[28],Perez和Yaylayan[24]在碳水化合物降解机理的基础上,提出了类似的四碳前体物如丁醛糖(aldotetrose)和2-脱氧丁醛糖(2-deoxy-aldotetrose)的形成,这两种物质能转化为呋喃(如图2)。另一方面,Becalski和Seaman[25]提出:在形成过程中存在中介物质2-糠酸(2-furoic acid),经脱羧后,形成呋喃。然而,以上两种观点是相符的,因为丁醛糖和2-脱氧丁醛糖的前体物在脱羧之前,可以通过环化生成呋喃环,然后形成2-糠酸(如图3)。由于适当标记的抗坏血酸存在无效性,所以这些机制仍然是建议并且需要进一步的实验验证。图3是根据Yaylayan V A[23]的文献报导修改而来的。
3.4 通过多不饱和脂肪酸的热氧化形成呋喃
Becalski和Seaman[25]通过研究发现,只有多不饱和脂肪酸,如亚油酸和亚麻酸经加热能形成呋喃。亚麻酸形成的呋喃是亚油酸的4倍多,并且三氯化铁催化可以使呋喃形成的量增加几倍。虽然亚油酸和亚麻酸的甘油三脂也可产生大量的呋喃,然而,在三氯化铁存在时,甘油三脂比游离酸生成更少的呋喃。这个发现不足为奇,因为5-戊基呋喃(5-pentylfuran)作为呋喃的一种衍生物,目前正作为酸败的一种化学标记物。5-戊基呋喃的产生与4-羟基-2-壬烯醛(4-HNE)的生成有关[29]。4-HNE的乙醇溶液在酸性条件下回流,可以形成呋喃。这种看法,在后来得到了证实。最近的研究发现5-戊基呋喃的浓度与橄榄油氧化时间呈显著正相关性[30]。一般情况下,多不饱和脂肪酸的氧化降解和脂质过氧化物的形成对生物系统中退化性疾病和食物的口味丧失及酸败的发生发挥重大作用。多不饱和脂肪酸通过活性氧的非酶化作用或脂氧合酶的酶解作用可以形成脂类氢过氧化物。随后,多不饱和脂肪酸的氢过氧化物在过渡金属离子的催化下,发生均裂,形成2-烯烃醛(2-alkenal)、4-氧代-2-烯烃醛(4-oxo-2-alkenal)和4-羟基-2-烯烃醛(4-hydroxy-2-alkenal)(见图4)。Perez和Yaylayan[24]提出呋喃(类似于5-戊基呋喃)可以由相应的4-羟基-2-丁烯(4-hydroxy-2-butenal)通过环化、脱水形成(见图4)。
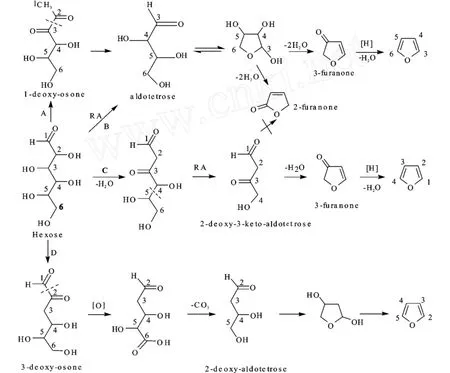
图2 通过己糖形成呋喃的途径Fig.2 Mechanistic pathways of formation of furan from hexose

图4 从多不饱和脂肪酸形成呋喃的途径Fig.4 Mechanistic pathways of formation of furan from oxidation of PUFA
4 食品中呋喃的检测方法
由于呋喃分子质量小,挥发性强,其定量容易受到复杂基质的干扰。根据已有文献,目前有关食品中呋喃检测的方法主要有两种:一是顶空进样-气相色谱-质谱法[31-34],另一种是固相微萃取-气相色谱-质谱法[35-37]。
4.1 顶空进样-气相色谱-质谱法
顶空进样是分析强挥发性化合物最合适的方法[38-39]。这种方法是通过顶空装置将样品中的呋喃提取出来,以D4-呋喃作为内标物,HP-PLOTQ石英毛细管柱作为分析柱,采用气相色谱分离,质谱定性定量[1]。
4.1.1 样品制备 由于呋喃的易挥发性,液上气体取样是呋喃分析的最好方法。为了减少损失,食品样品在处理前需要在4℃条件下冷却,并且需要在冰浴上用一个冷的搅拌器有效地进行均质化。纯的液体试样在加入内标物之前,称量后直接加入到顶空容器中;固体样品需要加冷水进行匀浆化。
4.1.2 分析条件
1)顶空条件:平衡温度90℃,平衡时间30 min,样品瓶低速振动;定量环温度100℃;传输线温度110℃,瓶加压时间0.5 min;样品压103.4 kPa(15psi);填充定量环时间0.5 min,定量环平衡时间0.1 min;进样时间1 min,GC循环时间35 min。
2)气相色谱条件:色谱柱:HP-PLOT Q石英毛细管色谱柱(30 m×0.32 mm×25μm);柱温升温程序:起始温度50℃,保持1 min后以10℃/min升温速率升至200℃,保持12.5 min;进样口温度: 200℃;载气:高纯氮气,流量1.7 mL/min;不分流进样,分流阀开阀时间0.75 min。
3)质谱条件:离子源:电子轰击(EI)离子源,电子能量70 eV;离子源温度230℃;四极杆温度150℃;传输线温度225℃;全谱扫描,质量扫描范围m/z=25~150;溶剂延迟时间2.5 min。
4.1.3 定性及定量 分别选择m/z=68和m/z= 72作为呋喃和D4-呋喃的定量离子,以不同浓度的呋喃和D4-呋喃的峰面积比与两者的质量比作标准曲线,以保留时间和68/39、72/42两对离子的响应强度比例分别作为呋喃和D4-呋喃的定性标准,实际样品中各对离子的强度比不超过标准样品溶液的±20%。
4.2 固相微萃取-气相色谱-质谱法
固相微萃取技术[40-41](SPME)是在固相萃取技术基础上发展起来的一种新的萃取分离技术,由加拿大WATEROO大学、美国SUPELCO公司和美国VARIAN公司联合开发。目前由美国SUPELCO公司生产手柄式固相微萃取装置,美国VARIAN公司生产气相色谱仪自动进样器式的微固相萃取装置。
固相微萃取-气相色谱-质谱法与顶空进样-气相色谱-质谱法的主要不同之处是进样方式的不同,固相微萃取试验是用萃取纤维头进行的,SPME纤维头上薄膜由极性的聚丙烯酸酯、聚乙二醇或非极性的聚二甲基硅氧烷组成。使用SPME时,先使纤维头缩进不锈钢管内,使不锈钢针管穿过盛装待测样品瓶的隔垫,插入瓶中并推手柄杆使纤维头伸出针管,纤维头可以浸入待测样品中或置于样品顶空,待测有机物吸附于纤维涂膜上,通常2~30 min吸附达到平衡,缩回纤维头,然后将针管推出样品瓶。最后,将SPME针管插入GC进样器,被吸附物经热解吸后进入气相色谱柱,开启流动相通过解吸池洗脱样品进样。其后的GC-MS分析条件与
4.1节所述方法相同,不再赘述。
目前,国内外出现的文献报道多数是以上述两种方法之一来检测食品中呋喃的含量。相对于固相萃取而言,顶空进样法在呋喃检测方面的优势在于经济快速,样品前处理过程简单,而且可以完成大量样本的自动分析,故应用更为广泛。
5 食品中呋喃的研究展望
目前,国外关于食品中污染物的研究比较系统,有关食品中呋喃的毒理学、前体物质和形成途径以及检测方法等都作了较为广泛的研究,但尚不完善,还有待于进一步深入。在呋喃的毒理学研究方面,还需进一步对呋喃致生殖和发育毒性进行研究,需要弄清呋喃对人体的致癌性到底有多大危害以及相应的致癌机理,以得到呋喃完整的毒性数据;在呋喃的前体物质和形成途径方面,还需进一步建立相应模型,对呋喃形成机制及其影响因素动力学进行研究,以得到食品加工过程中呋喃的安全控制新理论和新方法;在呋喃的分析检测上,还需进一步完善相关方法,建立更为有效、快捷的检测技术,进而对我国更多的食品进行检测,从而为研究我国食品中呋喃的暴露量并提出其限量标准以及为世界卫生组织对该污染物的评价提供科学依据。总之,为了填补我国在这一领域的研究空白和保护公众健康,非常有必要对食品中的呋喃进行更为深入广泛地研究。
[1]刘平,薛颖,金庆中,等.顶空气相色谱-质谱法测定婴幼儿食品中的呋喃[J].色谱,2008,26(1):35-38.
LIU Ping,XUE Ying,J IN Qing-zhong,et al.Determionation of furan in baby foods using headspace gas chromatographymass spectrometry[J].Journal of Chromatography,2008,26(1):35-38.(in Chinese)
[2]International Agency for Research on Cancer,WHO.IARC Monographs on the Evaluation of Carcinogenic Risks to Humans,1995,63:393.
[3]Health Canada.Fact sheet:furan in foods[EB/OL].(2004-10-01).http://www.hc-sc.gc.ca/fn-an/security/chem.-chim/furan/index_e.html
[4]Forsyth D S,Becalski A,Casey V,et al.Furan:mechanisms of formation and levels in food[EB/OL].(2004-04-04). http://www.fda.gov
[5]US Food and Drug Administration,Office of Plant and Dairy Foods.Exploratory data on furan in food[EB/OL].(2004) http://www.cfsan.fda.gov.
[6]EFSA.Report of the scientific panel on contaminants in the food chain on provisional findings of furan in food[J].EFSA Journal,2004a,137:1-20.
[7]EFSA.Report of the CONTAM panel on provisional findings on furan in food.annexe corrigendum[EB/OL].2004b,http://www.efsa.europa.eu.
[8]Maga J A.Furans in foods[J].Critical Reviews in Food Science and Nutrition,1979,4,355-400.
[9]Crews L,Castle A.A review of the occurrence,formation and analysis of furan in heat-processed foods[J].Trends in Food Science and Technology,2007,18:365-372.
[10]NTP.Toxicology and carcinogenesis studies of furan(CAS No.110-00-9)in F344/N rats and B6C3Fl mice(gavage studies)[R].NC:U.S.Department of Health and Human Services,Public Health Service,1993.
[11]Kedderis GL,Carfagna M A,Held SD,et al.Kinetic analysis of furan biotransformation by F-344 rats in vitro[J].Toxicol Appl Pharmacol,1993,123:274-282.
[12]金庆中,刘平,孟媛,等.北京市售婴幼儿食品中呋喃污染状况及相关暴露量的现况研究[J].卫生研究,2008,37(4): 471-473.
J IN Qing-zhong,LIU Ping,MENG Yuan,et al.Contaimination status and relatively exposure of furan on Beijing infant food[J].Journal of Hygiene Research,2008,37(4):471-473.(in Chinese)
[13]Burka L T,Washburn K D,Irwin R D.Disposition of[14C]-furan in the male F344 rat[J].Journal of Toxicology and Environmental Health,1991,34(2):245-257.
[14]Chen L J,Hecht S S,Peterson L A.Identification of cis-2-butene-1,4-dial as a microsomal metabolite of furan[J]. Chemical Research in Toxicology,1995,8(7):903-906.
[15]Byrns M C,Predecki D P,Peterson L A.Characterization of nucleoside adducts of cis-2-butene-1,4-dial,a reactive metabolite of furan[J].Chemical Research in Toxicology,2002,15(3):373-379.
[16]Lee H,Bian S S,Chen YL.Genotoxicity of 1,3-dithiane and 1,4-dithiane in the CHO/SCE assay and the Salmonella/ microsomal test[J].Mutation Research,1994,21(4):213-218.
[17]McGregor D B,Brown A,Cattanach P,et al.Responses of the L5178Y tk+/tk-mouse lymphoma cell forward mutation assay:mouse lymphoma cell forward mutation assay.III.72 coded chemicals[J].Environmental and Molecular Mutagene-sis,1988,12:85-154.
[18]Peterson L A,Naruko K C,Predecki D P.A reactive metabolite of furan,cis-2-butene-1,4-dial,is mutagenic in the ames assay[J].Chemical Research in Toxicology,2000,13(7):531-534.
[19]Kedderis GL,Ploch S A.The biochemical toxicology of furan[J].CIIT(Chemical Industry Institute of Toxicology)Activities,1999,19(12):1-10.
[20]Mugford C A,Carfagna M A,Kedderis GL.Furan-mediated uncoupling of hepatic oxidative phosphorylation in Fischer-344 rats[J]:an early event in cell death.Toxicology and Applied Pharmacology,1997,144(1):1-11.
[21]Louise J K,Kettil S,Lilianne A Z.Furan is not genotoxic in the micronucleus assay in vivo or in vitro[J].Toxicology Letters,2007,169:43-50.
[22]Baltes W,Bochmann G.Model reactions on roast aroma formation.1.Reaction of serine and threonine with sucrose under the conditions of coffee roasting and identification of new coffee aroma compounds[J].Journal of Agricultural and Food Chemistry,1987,35(3):340-346.
[23]Yaylayan V A.Precursors,formation and determination of furan in food[J].Journal Verbr Lebensm,2006,1:5-9.
[24]Perez Locas C P,YAYLAYAN V A.Origin and mechanistic pathways of formation of the parent furan-a food toxicant [J].Journal of Agricultural and Food Chemistry,2004,55(22):6830-6836.
[25]Becalski A,Seaman S.Furan precursors in food:A model study and development of a simple headspace method for determination of furan[J].Journal of AOAC International,2005,88:102-106.
[26]US FDA.Question and answers on the occurrence of furan in food[EB/OL].(2004-06-30)http://www.cfsan.fda.gov/~dms/furanqa.html
[27]Limacher A,Kerler J,Davidek T,et al.Formation of furan and methyl-furan by Maillard-type reactions in model systems and food[J].Journal of Agricultural and Food Chemistry,2008,56:3639-3647.
[28]LIAO M L,SEIB P A.Selected reactions of L-ascorbic acid related to foods[J].Food Technology,1987,41:104-107, 111.
[29]Sayre L M,Arora P K,Iyer R S,et al.Pyrrole formation from 4-hydroxynonenal and primary amines[J].Chem Res Toxicol,1993,6:19-22.
[30]Vichi S,Pizzale L,Conte L S,et al.Solid-phase micro-extraction in the analysis of virgin olive oil volatile fraction:Modifications induced by oxidation and suitable markers of oxidative status[J].Journal of Agricultural and Food Chemistry, 2003,51:6564-6571.
[31]Reinhard H,Sagar F,Zoller O,et al.Furan in foods on the Swiss market-Method and results[J].Mitt Lebensm Hyg, 2004,95:532-535.
[32]Becalski A,Forsyth D,Casey V,et al.Development and validation of a headspace method for determination of furan in food[J].Food Additives and Contaminants,2005,229:535-540.
[33]Sebyuva H Z,Goekmen V.Analysis of furan in foods:Is headspace sampling a fit-for-purpose technique?[J].Food Additives and Contaminants,2005,22:198-202.
[34]Nyman P J,Morehouse K M,Mcneal T P,et al.Single-Laboratory Validation of a Method for the Determination of Furan in Foods by Using Static Headspace Sampling and Gas Chromatography/Mass Spectrometry[J].Journal of AOAC International,2006,89(5):1417-1424.
[35]Goldmamann T,Perisset A,Scanlan F,et al.Rapid determination of furan in heated foodstuffs by isotope dilution solid phase micro-extraction-gas chromatography mass spectrometry(SPME-GC-MS)[J].Analyst,2005,130:878-883.
[36]Ho I P,Yoo S J,Tefera S.Determination of furan levels in coffee using automated solid-phase micro-extraction and gas chromatography/mass spectrometry[J].Journal of AOAC International,2005,88:574-576.
[37]Bononi M,Tateo F.Determination of furan by headspace solid-phase micro-extraction gas chromatography-mass spectrometry in balsamic vinegars of Modena(Italy)[J].Journal of Food Composition and Analysis,2008:1-14.
[38]Tassan C G,Russell G F.Sensory and gas chromatographic profiles of coffee beverage headspace volatiles entrained on porous polymers[J].Journal of Food Science,1974,39:64.
[39]Yang X,Peppard T.Solid-phase micro-extraction for flavor analysis[J].Journal of Agricultural and Food Chemistry, 1994,42:1925-1930.
[40]Bianchi F,Careri M,Mangia A,et al.Development and validation of a solid micro-extraction-gas chromatography-mass spectrometry method for the determination of furan in baby-food[J].Journal of Chromatography A,2006,1102:268-272.
[41]陈家华,方晓明,朱坚,等.现代食品分析新技术[M].北京:化学工业出版社,2004,21-23.
(责任编辑:朱明)
Recent Progress of Furan in Heat-Processed Foods
XIE Ming-yong, HUANGJun-gen, NIE Shao-ping
(State Key Laboratory of Food Science and Technology,Nanchang University,Nanchang 330047,China)
In recent years,food safety has attracted more and more attention.In this review,the research progress of the occurrence,toxicology,formation paths and detection technology of furan in heat-processed foods were summarized and comprehensively introduced.The analysis and comments of the progress was also supplied in this review manuscript.
furan,heat-processed foods,formation paths,detection technology
O 626.11
:A
1673-1689(2010)01-0001-08
2009-10-15
国家自然科学基金项目(30960242);教育部长江学者和创新团队发展计划项目(IRT0540)。
谢明勇(1957-),男,江西宜春人,教授,博士生导师,主要从事食品化学与营养学、食品安全研究。Email:myxie@ncu.edu.cn
猜你喜欢
山东化工(2020年5期)2020-04-07
山西大同大学学报(自然科学版)(2016年6期)2016-01-30
烟草科技(2015年8期)2015-12-20
食品工业科技(2015年6期)2015-10-21
哈尔滨工业大学学报(2015年8期)2015-09-04
天津科技(2015年11期)2015-06-26
湖南师范大学自然科学学报(2015年2期)2015-02-27
应用化工(2014年11期)2014-08-16
无机化学学报(2014年6期)2014-02-28
丝绸(2014年1期)2014-02-28
