
GC法检测培美曲塞二钠原料药中4种有机溶剂残留量
2010-08-07 01:25:54蔡东向池秀珍福建晋江市医院药剂科晋江市3600福建泉州市药品检验所泉州市36000
中国药房 2010年41期
蔡东向,池秀珍(1.福建晋江市医院药剂科,晋江市3600;.福建泉州市药品检验所,泉州市36000)
培美曲塞二钠是抗肿瘤药,在该原料药的制备中使用了N,N-二甲基甲酰胺(DMF)、二氯甲烷、乙醇、丙酮等有机溶剂,其中二氯甲烷和DMF为第2类溶剂,限量分别为0.06%、0.088%,乙醇和丙酮为第3类溶剂,限量均为0.5%[1]。笔者在对该药进行分析时发现二氯甲烷和丙酮在聚乙二醇-20M(PEG-20M)毛细管柱系统下难以分离,而在二甲基聚硅氧烷(DB-1)毛细管柱系统下丙酮与乙醇难以分离,考虑到各种溶剂的限量差异,故采用对4种溶剂两两分别分析测定的气相色谱(GC)法检测4种溶剂的残留量,结果表明所建立的方法简单、准确、灵敏度高、重复性好。
1 仪器与试药
GC-14B气相色谱仪、氢火焰离子化检测器(FID)、N-2000色谱工作站(浙江大学信息工程研究所)。
培美曲塞二钠原料药(北京思普润安医药科技有限公司,批号:20041002、20041003、20041004,含量:98.92%、99.28%、99.06%);二氯甲烷、DMF、四氢呋喃(THF)、无水乙醇、丙酮均为分析纯,水为重蒸水。
2 方法与结果
2.1 二氯甲烷和DMF的检查
2.1.1 色谱条件。色谱柱:DB-1石英毛细管柱(30 m×0.53 mm,1.0 μm);柱温:40℃;气化室:180℃;检测器:200℃;载气:氮气(N2):50 kpa,氢气(H2):50 kpa,空气:50 kpa;进样量:1 μL(不分流进样);溶剂为水。
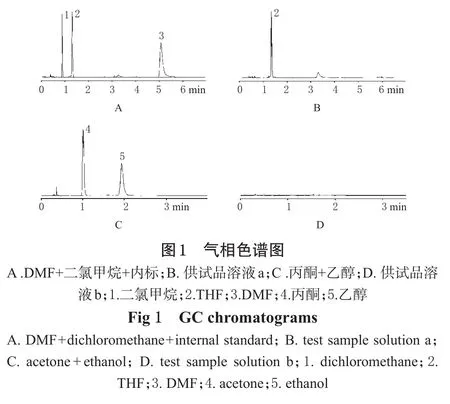
在上述色谱条件下,取“2.1.2”项下二氯甲烷、DMF和内标THF的混合标准溶液进样,记录色谱,理论板数按二氯甲烷计不得低于4000,二氯甲烷、DMF及THF两两之间的分离度应大于1.0。色谱见图1A。
2.1.2 溶液的制备。(1)标准贮备液:分别精密称取二氯甲烷60 mg、DMF 88 mg置于同一100 mL容量瓶中,加水定容至刻度,摇匀,即得。(2)内标溶液:精密称取THF 75 mg置于100 mL容量瓶中,加水定容至刻度,摇匀,即得。(3)对照品溶液:精密量取标准贮备液及内标溶液各1.00 mL,置于10 mL容量瓶中,加水定容至刻度,摇匀,即得。(4)供试品溶液a:精密称取1 g的样品,置于10 mL容量瓶中加水溶解,加1 mL内标溶液,加水定容至刻度,摇匀,即得。
取供试品溶液a进样,色谱见图1B。
2.1.3 标准曲线的制备。精密量取标准贮备液0.2、0.5、1.0、2.0、3.0 mL,分别置于10 mL容量瓶中,每瓶中加1 mL内标溶液,加水定容至刻度,摇匀,即得标准系列溶液。分别吸取上述标准溶液1 μL进样(每点3次),按“2.1.1”项下色谱条件进行分析,以待测溶剂与内标峰面积比值(Y)对浓度(X)进行回归,计算线性方程,得二氯甲烷的回归方程为Y=0.0166X-0.0911(r=0.9988);DMF的回归方程为Y=0.0334X-0.1669(r=0.9997)。结果表明,二氯甲烷、DMF的检测浓度线性范围分别为11.4~171、14.76~221.4 μg·mL-1。
2.1.4 检测限。取标准曲线最低点的标准溶液,用水稀释10倍,调整进样量至响应信号峰高为基线的3倍,即为检测限。测得二氯甲烷和DMF的检测限分别为0.30、0.02 ng。
2.1.5 精密度试验。取“2.1.3”项下的第3号溶液,精密吸取1 μL,分别进样5次,记录峰面积,以峰面积比值计算日内精密度,结果二氯甲烷、DMF的RSD分别为1.4%、1.9%;另取该溶液分别于3 d内进样,以峰面积比值计算日间精密度,结果RSD分别为1.7%、2.2%。
2.1.6 回收率试验。取已知残留量的样品1 g,精密称定,置于10 mL容量瓶中,加水适量溶解,精密加入标准贮备液及内标溶液各1.00 mL,加水定容至刻度,摇匀,得样品溶液。分别取样品溶液及“2.1.2”项下对照品溶液各1 μL,注入气相色谱仪,以峰面积按内标法计算回收率,结果见表1。
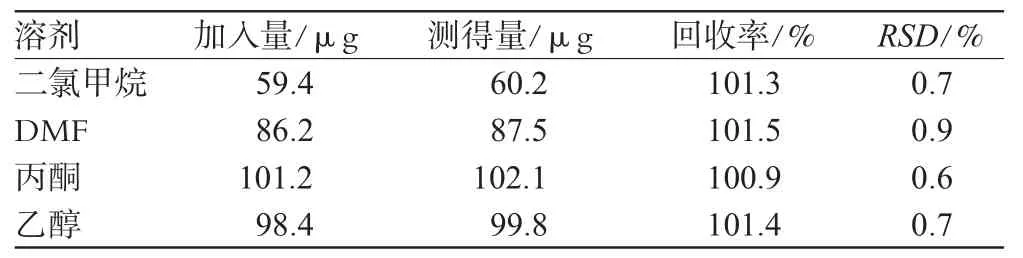
表1 回收率试验结果(n=3)Tab 1Results of recovery test(n=3)
2.1.7 样品中二氯甲烷和DMF的残留量测定。精密称取1 g的样品,置于10 mL容量瓶中加蒸馏水溶解,加1 mL内标溶液,加水定容至刻度。精密吸取1 μL进样,记录峰面积,结果二氯甲烷和DMF均未检出,说明样品符合要求。
2.2 丙酮和乙醇的检查
2.2.1 色谱条件。色谱柱:PEG-20M石英毛细管柱(30 m×0.53 mm,1.0 μm);柱温:42℃;气化室:180℃;检测器:200℃;恒温载气:N2:50 kpa,氢气:50 kpa,空气:50 kpa;进样量:1 μL(不分流进样);溶剂为水。
在上述色谱条件下,取“2.2.2”项下乙醇与丙酮的标准溶液进样,各组分色谱峰的理论板数均不得低于3000,二者分离度应大于2.0,色谱见图1C。
2.2.2 溶液的制备。(1)标准贮备液:精密称取丙酮和乙醇各0.1 g置于同一100 mL容量瓶中,加水定容至刻度,摇匀,即得。(2)对照品溶液:精密量取标准贮备液1.00 mL,置于10 mL容量瓶中,加水定容至刻度,摇匀,即得。(3)供试品溶液b:取样品,精密称取1 g置于10 mL容量瓶中,以水稀释至刻度,摇匀,即得。
取供试品溶液b进样分析,色谱见图1D。
2.2.3 标准曲线的制备。精密量取0.2、0.5、1、2、3 mL标准贮备液,分别置于10 mL容量瓶中,加水定容至刻度,摇匀,即得标准系列溶液。分别吸取上述溶液1 μL进样(3次)分析,记录峰面积,以各浓度点峰面积平均值(Y)对浓度(X)进行回归,得乙醇的回归方程为Y=701.77X+1562.6(r=0.9996),丙酮的回归方程为Y=677.46X-997.3(r=0.9990)。结果表明,乙醇与丙酮的检测浓度线性范围均为20~300 μg·mL-1。
2.2.4 检测限。取标准曲线最低点浓度的标准溶液用水稀释50倍进样,调整进样量至响应信号峰高为基线的3倍即为检测限。按此方法测得乙醇和丙酮的检测限分别为0.09、0.02 ng。
2.2.5 精密度试验。取“2.2.3”项下第3号溶液,精密吸取1 μL,分别进样5次,记录峰面积,以峰面积计算日内精密度,结果乙醇和丙酮的RSD分别为0.4%、1.6%;另取该溶液分别于3 d内进样,以峰面积计算日间精密度,结果分别为0.9%、1.2%。
2.2.6 回收率试验。取已知残留量的样品1 g,精密称定,置于10 mL容量瓶中,加水适量溶解,精密加入标准贮备液1.00 mL,加水定容至刻度,摇匀,得样品溶液。分别取样品溶液及“2.2.2”项下对照品溶液各1 μL,注入气相色谱仪,以峰面积按外标法计算回收率,结果见表1。
2.2.7 样品中乙醇和丙酮残留量测定。分别精密吸取1 μL供试品溶液和对照品溶液进样,记录峰面积。结果乙醇和丙酮均未检出。
3 讨论
本品在合成过程中所用的4种有机溶剂在测定时,由于柱长有限,因而互相之间易产生干扰,如以SE-30(甲基硅橡胶)为固定液,则乙醇和丙酮不易分开;而以PEG-20M为固定液,则二氯甲烷与乙醇互相干扰。但分别测定时,则干扰可消除。考虑到DMF在PEG-20M柱上出峰较晚,故测定样品时采用程序升温(42℃保持3 min后,以15℃·min-1升至140℃并保持3 min),以保证DMF完全流出色谱柱。同时该色谱体系下的测定结果也表明,在SE-30体系中保留时间5.5 min处的峰为溶剂峰,而不是样品中的DMF残留。结果,样品中4种溶剂未检出。
人用药品注册技术规范国际协调在关于有机溶剂残留量的指导原则中将二氯甲烷、DMF列为第2类必须控制的毒性试剂,将乙醇、丙酮列为第3类低毒性的溶剂[2]。根据《化学药物残留溶剂研究的技术指导原则》[3],本品在水中易溶,同时残留的有机溶剂也能在水中溶解,所以采用水作为溶剂进行试验。本试验采用内标法测定有机溶剂的含量,可减少仪器系统或操作过程的误差。经对多种有机溶剂的筛选,选择THF为内标可与4种有机溶剂达到基线分离,无干扰,方法学试验得到满意结果,精密度也符合2010年版《中国药典》[1]的要求(RSD<5%)。故该法可用于测定培美曲塞二钠原料药中有机溶剂的残留量,测定结果准确、可信。
[1]国家药典委员会编.中华人民共和国药典(二部)[S].2010年版.北京:中国医药科技出版社,2010:附录61~65、194.
[2]周海均主译.药品注册的国际技术要求·质量部分[M].北京:人民卫生出版社,2000:79.
[3]国家药品审评中心.化学药物残留溶剂研究的技术指导原则[Z].2005.
猜你喜欢
上海化工(2018年10期)2018-10-31 01:21:06
食品界(2016年4期)2016-02-27 07:37:06
大连工业大学学报(2015年4期)2015-12-11 04:06:50
电源技术(2015年7期)2015-08-22 08:48:52
电测与仪表(2015年7期)2015-04-09 11:40:30
应用化工(2014年4期)2014-08-16 13:23:09
电测与仪表(2014年9期)2014-04-15 00:27:16
电测与仪表(2014年11期)2014-04-04 09:21:40
化工生产与技术(2014年6期)2014-02-27 13:42:07
化工生产与技术(2014年5期)2014-02-27 13:42:02
