气相色谱法测定酒剂及蒸馏酒中乙醇量
2010-07-29 08:21肖顺经曾庆艳倪兆武
中国药业 2010年18期
肖顺经,曾庆艳,倪兆武,曾 波
(云南省昭通市食品药品检验所,云南 昭通 657000)
酒剂、蒸馏酒及配制酒中乙醇量的测定方法各不相同,药典中酒剂的乙醇量测定方法为气相色谱法或蒸馏法(相对密度法)[1],《食品卫生检验方法》中蒸馏酒及配制酒的乙醇量测定方法为蒸馏法(比重计法)[2]。相比之下,气相色谱法更方便、快捷、精确。笔者参照《蒸馏酒及配制酒卫生标准的分析方法》,在药典中气相色谱法和乙醇量测定法的基础上,采用气相色谱法测定了酒剂及蒸馏酒中乙醇量,报道如下。
1 仪器与试药
GC-14Bpf型气相色谱仪(日本岛津)。白酒(市售,酒精度50%);藿香正气水(云南腾冲制药厂,批号为071110,080111,080208,080501;云南云河药业有限公司,批号为070401;云南天利药业有限责任公司,批号为20080202);无水乙醇(分析纯,成都市联合化工试剂研究所),正丙醇(分析纯,北京化学试剂公司),纯净水。
2 方法与结果
2.1 色谱条件
色谱柱:GDX-102填充柱(2 m);柱温:140℃;FID检测器温度:190℃;进样口:不分流,温度170℃;载气:氮气,流量100 mL/min;燃气:氢气,流量60 mL/min;助燃气:空气,流量500 mL/min;进样量:0.5 μL 。
2.2 系统适用性试验
精密量取无水乙醇4,5,6 mL,分别置100 mL量瓶中,分别精密加入正内醇5 mL,加水至刻度,摇匀,得校正因子溶液。取无水乙醇、正丙醇、校正因子溶液各0.5 μL,注入气相色谱仪,分别连续进样 3次,测定峰面积。结果校正因子(f)为 1.26,RSD为 0.71%(n=9)。正丙醇理论板数大于1000,乙醇和正丙醇两峰的分离度大于4(见图1)。
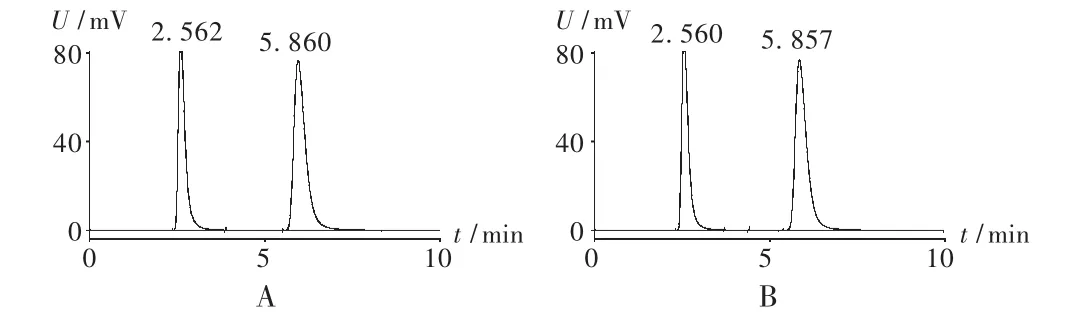
图1 气相色谱图
2.3 方法学考察
保留时间及检出限试验:取无水乙醇和正丙醇各5 mL,分别置100 mL量瓶中,用水稀释至刻度,摇匀,进样0.5 μL,分别测定6次。无水乙醇保留时间为2.546 min,RSD为0.84%(n=6),检出限为0.008 ng;正丙醇保留时间为 5.821 min,RSD 为 0.74%(n=6),检出限为 0.006 ng。
内标物质试验:取无水乙醇和供试品分别直接进样0.5 μL,连续3次。结果在正丙醇保留时间处无对应峰。
线性关系考察:精密量取无水乙醇对照品适量,加纯净水制成每1 mL 含 50 μL 的溶液,精密量取 1.0,2.0,4.0,6.0,8.0,10.0 mL,分别置100 mL量瓶中,加纯净水至刻度,摇匀,进样,记录色谱图。以体积浓度为横坐标、峰面积为纵坐标绘制标准曲线,得回归方程y=3.012 x+0.822,r=1.0000(n=6)。结果表明,无水乙醇体积浓度在2~7 μL/mL范围内与峰面积线性关系良好。
稳定性试验:取同一供试品溶液,分别在 0,1,2,4,6,18 h 时进样测定,记录峰面积。结果的 RSD为0.24%(n=6),表明供试品溶液至少在18 h内稳定。
重现性试验:取同一批号样品5份,按供试品溶液制备方法制备溶液并测定方法测定乙醇量。结果藿香正气水的乙醇量为41.48%,RSD 为 0.21%(n=5)。
加样回收试验:精密量取已知含量(44.14%)的样品5份,加无水乙醇对照品溶液,用上述方法和内标法测定,计算回收率。结果见表1。

表1 无水乙醇加样回收试验结果(n=6)
2.4 样品含量测定
精密量取供试品10 mL,置100 mL量瓶中,精密加入正内醇5 mL,用水稀释至刻度,摇匀,得供试品溶液。精密吸取供试品溶液0.5 μL,连续进样 3 次,按峰面积计算样品乙醇量。结果见表2。
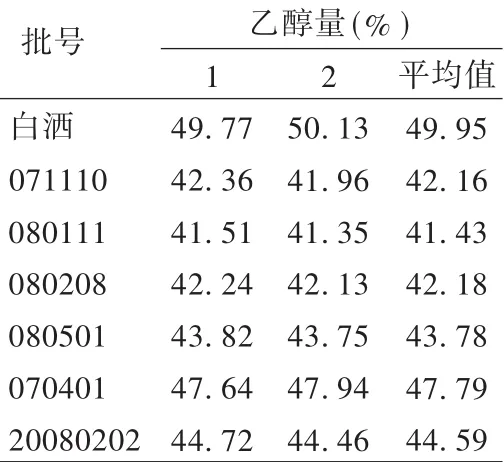
表2 样品乙醇量测定结果
3 讨论
藿香正气水为常备中成药,具有解表化湿、理气和中作用,主要用于外感风寒、内伤湿滞或夏伤暑湿所致的感冒等,其中乙醇作溶剂使用,同时具有佐使作用,故测定藿香正气水和白酒中的乙醇量具有代表性。本试验采用恒温分析方法,8 min就完全出峰,效果满意;值得提醒的是,乙醇检出限低,进样量不宜过大。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2005:21.
[2]GB/T5009.48-2003,中华人民共和国国家标准·食品卫生检验方法理化部分(一)蒸馏酒及配制酒卫生标准的分析方法[S].
猜你喜欢
中老年保健(2022年6期)2022-11-25
发明与创新(2022年31期)2022-11-03
当代水产(2022年4期)2022-06-05
中国医药科学(2022年5期)2022-05-05
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
养生保健指南(2019年3期)2019-12-16
中国继续医学教育(2015年6期)2016-01-07
天津医科大学学报(2015年2期)2015-12-22