
奶牛单基因遗传缺陷研究进展
2010-06-09 05:41周月君
中国牛业科学 2010年5期
周月君
(西北农林科技大学动物科技学院,陕西杨凌712100)
家畜遗传缺陷是指由于遗传物质变异对家畜个体造成的有害影响,表现为身体结构缺陷或功能障碍。在过去50年里,由于有效地应用基于数量遗传学的育种方案,家畜生产力得到了大幅的提高。高强度的选择,加上人工授精技术的普及和应用,极大地增加了家畜养殖的效益,但随之而来的是,畜群的近交系数逐渐增大,畜群有效含量逐渐减小。全世界存在有几亿头奶牛,而群体有效含量只有几百头,奶牛业面临着遗传缺陷病的定期暴发。
由于遗传物质变异对家畜个体造成的有害影响,表现为身体结构缺陷或功能障碍,这种有害的遗传信息按照一定的遗传方式在世代间垂直传递,在某些品种的家系内呈现一定的表达方式,不会延伸到无亲缘关系的个体中。家畜育种中,尤其是在以人工授精技术广泛应用的奶牛育种中,识别并淘汰造成遗传病的有害基因尤为重要。
1 遗传缺陷的危害
有效群体数的降低导致近交程度的增加,使得畜牧业面临着隐性缺陷病的定期暴发。例如,在上世纪90年代,14%的公牛携带编码白细胞 整合素亚单位CD18基因的突变,从而引起白细胞黏附缺陷(BLAD)。这一致死的免疫缺陷使得0.2%的新生犊牛死亡,估计每年给美国造成的经济损失为5亿美元。而近期发现25%的公牛是脊椎畸形综合症(CVM)的携带,该突变基因纯合时造成93%的母牛流产,给世界奶牛业造成了巨大损害。由于大多数遗传缺陷病均为隐性遗传,而携带者表现正常,因此很难发现。当某种遗传缺陷被发现时,其已经在畜群中进行了长时期的传播,这给养殖带来巨大的损失。如CVM 的最初携带者出生在1974年,是当时一头非常优秀的种公牛,但发现该病的时间是2000年,这已经过了26年,其所造成的直接经济损失是巨大的。而遗传缺陷病一旦发生,对疾病的诊断和控制、隐性携带者的检出和淘汰、育种方向的改变及育种计划的调整等都需要花费大量的人力和物力,造成不可估计的间接经济损失。而有害基因污染已有优良基因库,由此造成损失更不容忽视。
2 单基因遗传病的发病机理及遗传规律
遗传缺陷病的发病机理是由于调节机体内发育及代谢通路的遗传物质发生变异,使通路的某个环节受阻,从而引进机体结构异常或功能的损害。如白细胞粘附缺陷症(Bovine Leukocyte Adhesion Deficiency,BLAD)是一种牛的造血系统遗传性疾病,以严重的重复性感染、缺少脓液形成、损伤愈合延迟和白细胞增多症为特症,实质是白细胞粘附及相关的功能包括吞噬和趋化作用的缺陷。遗传缺陷具有先天性和家系遗传的特症,遵循孟德尔遗传规律向后代传递。对于占大部分的隐性遗传缺陷而言,由于等位基因(Aa)的显隐性关系,杂合子不会表现发病。但如果表型正常的携带者(Aa)交配产生表型正常与缺陷纯合个体的比为3∶1,后代基因型AA 、Aa、aa个体的比例为 1∶2∶1,也就是 50%个体为携带者,如果在胚胎时期aa个体流产或出生时即死亡,那么后代携带者为2/3,也就是67%的个体为携带者。其它的遗传方式有显性、不完全显性和超显性,同样可用孟德尔理论进行解释。
3 奶牛常见的遗传缺陷
到目前已报道的牛遗传缺陷共有376种,其中鉴定为单基因遗传的共有75种,已阐明分子机制的有43种,作为人类疾病潜在模型的126种。其他家畜遗传缺陷情况见表1。而国际荷斯坦牛联盟(World Holstein-Friesian Federation,WHFF)及加拿大荷斯坦奶牛协会要求在系谱标明的有6种遗传缺陷,包括牛白细胞粘附缺陷、脊椎畸形综合症、尿苷酸合酶缺陷症、并趾症(也称为螺蹄症)、瓜氨酸血症、凝血因子XI,其染色体位置及在系谱上的标识见表2。下面就以上6种遗传缺陷情况作一简述。
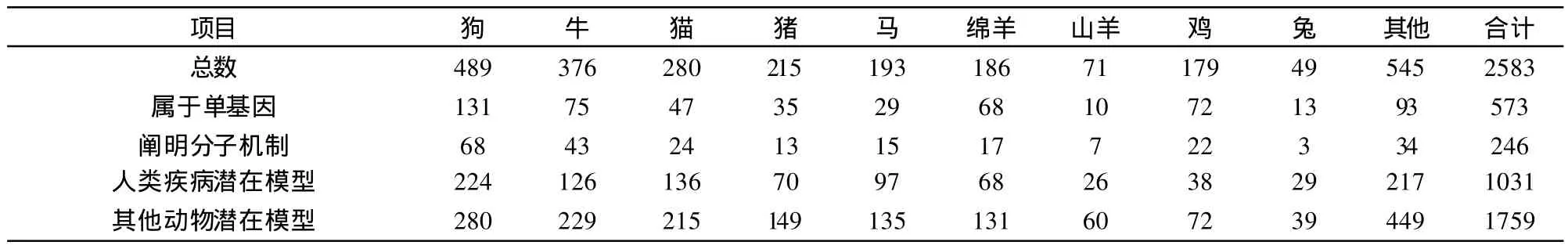
表1 家畜遗传缺陷汇总表
3.1 脊椎畸形综合症
脊椎畸形综合症(complex vertebral malformation,CVM)最早由丹麦科学家于2001年发现[1]。最初丹麦科学家发现荷斯坦牛群中存在脊椎畸形,两前腿筋腱变短、无法直立行走,颈短、心脏畸形等综合症状的新生病畜,并且发现牛群中流产率偏高。经DNA鉴定发现了一个隐性遗传缺陷基因,其纯合时可以造成妊娠母牛流产、死胎或犊牛出生后很快死亡。随后美国、加拿大、日本和欧洲一些国家也相继报道[2],随后建立了DNA分子检测方法。系谱研究发现,所有发病个体均可以追溯到美国一头非常著名的公牛“Carlin-M Ivanhoe Bell”(登记号:1667366),这头公牛可能是该遗传缺陷基因的共同祖先,出生在1974年,其父亲 Penstate Ivanhoe Star,出生在1963年,被认为是变异源,从系谱标识上看出,他们同时是BL的携带者。Carlin-M Ivanhoe Bell在当时因其极优良的生产性能而在全世界范围内被广泛使用,致使20几年后他所携带的隐性有害基因得到广泛传播。在种公牛中,美国、德国、瑞典、日本CVM携带者频率分别为17.76%,13.17%、23.25%和32.50%,英国排名前100名的公牛携带者频率为16%。我国,2006年初芹对68荷斯坦种公牛进行了检测发现10头携带者[3],计算频率为14.71%。脊椎畸形综合症造成胎儿的早期流产、死胎或早产,只有极少数个体出生时还能够存活一段时间,但很快死亡或被淘汰。据统计,在母牛妊娠的100 d内,患CVM 的胚胎的流产率是29%;当妊娠到150 d时,流产率上升到45%;当妊娠260 d时,有77%的CVM 胎儿已经死亡,而整个妊娠期能存活的CVM胎儿仅有7%,实际出生时犊牛的存活率可能更低[4,5]。CVM遗传缺陷的发现立刻引起了各国奶牛育种协会和育种工作者的重视。当前该遗传缺陷病已经得到了控制,如在加拿大2002年和2003年出生的犊牛携带频率为2.5%,当前荷斯坦母牛中携带频率为1.67%。

表2 6种遗传缺陷染色体位置及在系谱上的标识
3.2 白细胞粘附缺陷症
白细胞粘附缺陷症(bovine leukocyte adhesion deficiency,BLAD),最早在1983年发现,1984年确定病因,它是一种与白细胞粘附有关的细胞表面糖蛋白(整合素)的表达缺陷所致。多发生于1~14个月龄的荷斯坦牛,是常染色体单基因隐性遗传类型,双亲均可作为缺陷的携带者。出生小牛如为本病的纯合体,临床上即显症发病,表现为持续性或反复感染发炎、生长发育受阻、嗜中性粒细胞持续性增加等症状。Shuster等对1头患 BLAD的 Holstein牛CD18编码基因进行了序列分析,结果发现,383位的碱基由A变为G(A383G),与之相应的位于胞外高度保守区的128位的天门冬氨酸变成了甘氨酸,致使白细胞表面的整合素表达明显减少或缺乏而引起临床发病。另1个碱基则发生T775C变异,但其对应的氨基酸都是亮氨酸,故不发病。牛的CD18基因已经被定位在1号染色体上,GENBANK中牛CD18DNA序列全长为1640bp(Y12672)[6]。BLAD携带者的共同祖先是“Osborndale Ivanhoe”(注册号 :1189870),出生在 1952年,其儿子 “Penatate Ivanhoe star”(注册号:1441440),出生在 1963年,孙子“Carlin-M Ivanhoe Bell”(注册号:1667366),出生在1974,均为携带者,同时后两者还是CVM的携带者。美国2025头荷斯坦公牛中发现BLAD携带率为14.1%,其中TPI排名前100名的公牛BLAD携带率高达17.1%。据估测,美国每年可以出生1.6万头BLAD牛,每头母牛按300美元计,每年引起的经济损失可达50万美元。2002年,韩国对109头荷斯坦公牛进行了检测,发现 5头BLAD携带者[7]。2003年,乌拉圭在121头母牛中发现8头BLAD携带者,17头公牛中发现 2头 BLAD携带者[8]。2004年,波兰对5785头公牛进行了检测,其中198头为BLAD携带者,尽管之前曾采取了一定的措施,但仍发现了携带者[9]。同年,捷克检测了246头牛,没有发现BLAD携带者。在加拿大,1992年出生的牛携带者频率达到5%,当前牛群中BLAD携带频率为1.28%,已经得到了控制。
3.3 尿苷酸合酶缺乏症
牛尿苷酸合酶缺乏症(deficiency of uridine monophophate synthase,DUMPS),也称为单普病,是荷斯坦牛群中一种常染色体单基因隐性遗传疾病,可导致隐性纯合的后代胚胎早期死亡。Schwenger B(1993)等人发现杂合子C-末端405处的密码子存在一个点突变,原编码精氨酸的密码子CGA转变为终止密码子的 TGA[10]。突变基因的转录产物最终翻译成一段C-末端缺失76个氨基酸催化亚基的短蛋白,造成乳清酸转化为鸟苷酸的催化功能丧失,不能合成DNA、RNA所需的嘧啶核苷酸,从而导致胚胎在妊娠40~60 d时死亡,死亡率100%。UMP基因的cDNA序列全长为1869bp(NM 177508)。DUMPS携带者的共同祖先是“Skokie Sensation Ned”(注册号:1308101),出生在1957年。该病上世纪80年末期才被发现。在我国,张松(1999)应用PCR-RFLP方法对北京市奶牛中心良种场(91头)、北京市南郊杜庆牛场(82头)、北京市南郊德茂牛场(190头)共363头中国荷斯坦奶牛和乌鲁木齐种牛场的193头新疆褐牛以及北京市种公牛站的59枚公牛冻精进行了尿苷酸合酶(UMPS)基因分子水平的检测。结果检出了2头杂合母牛,杂合子占所检测母牛群的0.551%,此频率处于国外报道的DUMPS发生频率的范围(0.2%~2.5%)之内。在荷斯坦公牛和新疆褐牛中只检测到一种基因型,没有发现隐性突变基因的携带者。在加拿大当前DUMP的携带者频率为0.068%,基因频率很低,可以认为已经从牛群中根除。
3.4 瓜氨酸血症
瓜氨酸血症(citrullinemia,CN)是一种引起荷斯坦牛尿循环代谢紊乱的常染色体单基因隐性遗传缺陷病。发病个体由于缺乏尿素代谢过程中催化瓜氨酸生成精氨酸琥珀酸的关键酶-精氨酸琥珀酸合成酶,而导致瓜氨酸不能转变为精氨酸琥珀酸,引起机体内尿代谢紊乱。这一代谢紊乱的发生使体内代谢生成的瓜氨酸前体-氨无法转变为尿素,而无法排出体外,从而使对机体有毒物质的氨大量积蓄于组织及血液中,引起机体发病。由于母体可以排出胎儿代谢产生的氨,所以发病个体在胎儿期间或刚出生时表现正常,但出生后不久便表现出一系列临床症状,如精神沉郁、步态紊乱、抽搐、惊厥、失明、虚脱等,一般出生后5 d死亡,死亡率100%。该病是精氨酸琥珀酸合成酶基因的一个点突变造成的。精氨酸琥珀酸合成酶外显子5发生C-T的点突变,使密码子CGA转变为终止密码子TGA,从而其翻译产物-精氨酸琥珀酸合成酶变成了截短了的85个AA的蛋白质(正常为412个AA),而失去活性[11]。Dennis根据此原理利用限制性内切酶AvaII建立了检测遗传缺陷CN的 PCR-RFLP方法,之后Robinson利用该方法对美国荷斯坦牛进行了检测。澳大利亚荷斯坦牛群中CN携带频率曾经很高,1989年澳大利亚人工授精组织的13%公牛是遗传缺陷CN的携带者,所有携带者均与公牛“Linmack Kriss King”(注册号:CAN294213)有关,Linmack Kriss King出生在1959年,被认为是CN携带者的共同祖先,与同时代的其他公牛相比,具有极高的遗传潜能,尤其是乳脂量育种值高,受到育种者及生产者的广泛欢迎。这使CN在荷斯坦牛群中广泛传播。到80年代,澳大利亚主要育种公司的75%公牛含有Linmack Kriss King的血液。1900年澳大利亚禁止CN携带者公牛进入人工授精中心,使该病的发生率明显降低。在加拿大奶牛协会网上(www.cdn.ca),共查到公牛Linmack kriss king的后代452头,均出生在20世纪70~80年代,其中澳大利亚有63头,法国8头,英国 213头,爱尔兰40头,新西兰128头。以英国出生的后代最多,并且在英国其后代有两头公牛参加国际公牛遗传评定。在当前牛群CN携带者的频率已很低,有的国家甚至检测不到。
3.5 凝血因子XI缺陷症
凝血因子XI(Factor XI,FXI)是一种丝氨酸蛋白酶原,在传统的内源性凝血途径中,因子XI被活化的因子XII激活后,在钙离子存在下,被激活,从而引发凝血过程的进行。近来研究发现,因子也能被凝血酶激活,活化的因子XI促进凝血酶的大量产生,近一步激活被凝血酶激活的纤溶抑制物,而抑制纤溶系统,起到凝血和抗凝作用。FXI主要在内源性凝血的前期反应中对相关因子的激活起关键性的作用。缺陷个体一般无直接表现症状,间接症状包括:注射治疗时出血时间延长、血腥奶、贫血,与血友病有些相似。缺陷纯合及杂合个体可能表现低的产犊率和成活率,并且对疫病的易感性。酶活试验发现感染个体FXI活性比正常个体低10%。对奶牛养殖具有重大的经济影响。FXI缺陷病已经在人类、狗和牛中相继报道,最早是在1969年的美国,后来在加拿大和英国。FXI基因包括15外显子和14个内含子,定位在牛 27q14。Marron et al(2004)在荷斯坦牛群中发现感染个体FXI基因的第12外显子上存在一个76 bp的插入突变,导致一个终止密码子的出现,使翻释后的FXI因子缺乏蛋白功能区[12]。并对美国419个荷斯坦牛进行了检测,结果检测出 5个杂合子,突变频率为1.2%。Kunieda et al(2005)在日本黑牛群体中发现感染个体在第9外显了上存在一个15bp的插入突变[13]。目前在加拿大还没有进行分子检测,没有确定共同祖先。
3.6 并趾症
牛并趾症(syndactyly),也称为骡蹄症(Mulefoot,MF),是一种常染色体的隐性遗传病,但在不同品种中表现不同的外显率,感染者一般一只或多只蹄子均可表现骡蹄。最早报到在1967年,到目前已经在多个国家的许多个品种中报到,然而大部分的文献都是发生在美国荷斯坦牛中。早期的研究发现,并趾症携带者可多产2.1磅的乳脂,298磅的奶量。1996年Charlier用基因组范围内的标记进行IBD连锁分析将该基因定位到牛15号染色体上。Duchesne(2006)对36个患有骡蹄症的荷斯坦母牛进行了研究,发现所有感个体的低密度脂蛋白受体结合蛋白基因(Low Density Lipoprotein Receptorrelated Protein 4,LPR4)第33外显子发生了两个连续碱基的突变(C4863A,G4864T),导致LRP4蛋白的两个遗传密码发生发改变,改变了保守性的类表皮样生长因子蛋白的结构。Johnson(2006)在两头感染的安格斯牛LRP4基因的第37内含子上发现了一个不同的单碱基突变。Cord Drögemǜller(2007)在荷斯坦牛、德国西门塔尔牛和西门塔尔-夏洛来牛的 LRP4基因上发现4个新的突变[14]。骡蹄症携带者的共同祖先是“Wayne Spring Fond Apollo”(注册号:159582),出生在 1970 年,曾经是金牌公牛。系谱分析发现该公牛是“Osborndale Ivanhoe”(注册号:1189870)的孙子 ,而“Osborndale Ivanhoe”是BLAD携带者的共同祖先。在加拿大荷斯坦牛群中MF的携带者频率为0.364%。
总之,大量的研究发现,随着人们对高产的不断追求,动物对疾病的抵抗力减弱,如高产奶牛乳房炎的发生率增加;同时,新的遗传缺陷也在不断的出现,如Carole CHarlier(2008)利用覆盖全基因组的高密度的SNP芯片,对牛的5个遗传缺陷进行了研究,精细定位了其中的3个遗传缺陷:比利时兰牛先天性肌张力障碍I和II,意大利契安尼娜牛鱼鳞病胎儿,并阐明了分子机理[15]。有些遗传疾病还与生产性能紧密连锁,如Weaver综合症与奶牛产奶性状间显著相关。这使许多国家都把对奶牛的抗病性选择纳入育种规划中。
在奶牛育种中,应监测所出现的遗传缺陷,并收集发病个体的样品,定位并鉴定突变位点,寻找诊断的标记,利用这些标记避免风险选配,进而通过有效的策略来控制遗传缺陷病的暴发是至关重要的。在我国奶牛育种中也应加强这方面的工作,随着我国奶牛群体的不断扩大,检测牛群的缺陷基因并淘汰缺陷个体,尤其是对种公牛而言,更是具有十分重要的意义。
[1]Agerholm J S,Bendixen C,Andersen O,et al.Complex vertebral malformation in Holstein calves[J].Journal of Veterinary Diagnostic Investigation 2001,13:283-289.
[2]Duncan R B,Carrig C B,Agerholm J S,et al.Complex vertebral malformation in a Holstein calf:report of a case in the USA[J].Journal of Veterinary Diagnostic Investigation.2001,13:333-336.
[3]Qin Chu,Dongxiao Sun,Ying Yu,et al.Identification of com-plex vertebral malformation carriers in Chinese Holstein[J].J Vet Diagn Invest.2008,20:228-230.
[4]Nielsen U S,Aamand G P,Andersen O,et al.Effects of complex vertebral mal-formation onfertility traits in Holstein cattle[J].Livest Prod Sci,2003,79(2):233-238.
[5]M alher X,Beaudeau F,Philipot J M.Effects of sire and dam genotype for complex vertebral malformation(CVM)on risk of return-to-service in Holstein dairy cows and heifers[J].Theriogenology 65:1215-25,2006.
[6]Shuster D E,Bosworth B T,Kehrli M E.Sequence of the Bovine CD18-Encoding cDNA-Comparison with the Human and Murine Glycoproteins[J].Gene.1992,114:267-271.
[7]Lee Y K,Chang K W,Nam I S,et al.Studies on the detections of congenital genetic disorder in Holstein proven and candidate bulls[J].Journal of Animal Science and Technology.2002,44(3):279-288.
[8]Llambi S,Guevara K,Rincon G,et al.Frequency of leukocyte adhesion deficiency in a population of Holstein-Friesian cattle in U ruguay[J].Ars Veterinaria.Faculdade de Ciencias Agrariase Veterinarias.2003,19(1):52-56.
[9]Otwinowska-Mindur A,Zarnecki A.Prevalence of bovine leukocy te adhesion deficiency(BLAD)in Polish Black-and-White Bulls[J].Animal Science Papers and Reports.2004,22:Supplement 2,77-80.
[10]Schwenger B,Tammen I,Aurich C.Detection of the homozygous recessive genotype for deficiency of uridine monophosphate synthase by DNA ty ping among bovine embryos produced in vitro[J].Journal of Reproduction and Fertility 1994,100:511-514.
[11]T homsen P D,Nielsen J S.PCR Screening for Carriers of Hereditary Citrullinaemia in Danish Holstein-Friesian Bulls[J].Acta Veterinaria Scandinavica 1991,32:279-282.
[12]Marron B M,Robinson J L,Gentry P A,et al.Identification of a mutation associated with factor XI deficiency in Holstein cattle[J].Anim Genet.200435:454-6.
[13]Kunieda M,T suji T,Abbasi A R,et al.An insertion mutation of the bovine Fii gene is responsible for factor XI deficiency in Japanese black cattle[J].Mamm Genome.2005,16:383-9.
[14]Drö gemǜller C,Leeb T,Harlizius B,et al.Congenital sy ndacty ly in cattle:four novel mutations in the low density lipoprotein receptor-related protein 4 gene(LRP4)[J].BM C Genet 2007,8:5.
[15]Carole Charlier,Wouter Coppieters,Fre'de'ric Rollin,et al.Highly effective SNP-based association mapping and management of recessive defects in livestock[J].Nature genetics,2008,40:449-454.
猜你喜欢
贵州畜牧兽医(2022年6期)2022-12-29
现代临床医学(2022年4期)2022-09-29
中国奶牛(2022年7期)2022-08-12
中老年保健(2021年12期)2021-08-24
中西医结合肝病杂志(2020年2期)2020-10-27
三农资讯半月报(2020年17期)2020-09-26
中国畜牧杂志(2020年3期)2020-03-17
河南农业科学(2019年5期)2019-05-28
文苑(2018年20期)2018-11-15
学生天地(2018年30期)2018-10-17
