
液相色谱法纯化固相合成戈那瑞林
2010-06-05 01:45:44肖建国,阮振兴,王林鹏等
化学与生物工程 2010年9期
戈那瑞林是治疗前列腺癌等与雌激素相关疾病的药物。戈那瑞林为促性腺激素释放素,通过竞争结合垂体LH-RH的大部分受体,使LH和FSH的生成和释放呈一过性增强,但这种刺激的持续,会导致受体的吞噬、分解增多,受体数减少,垂体细胞反应下降,LH和FSH分泌能力降低,因而抑制卵巢雌激素的生成。通过这种负反馈作用抑制垂体功能,从而起到治疗作用。连续使用可使LH和LH-RH分泌减少。戈那瑞林属于十肽化合物,其结构与天然提取物完全相同。其肽链结构为:Glu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2[1]。目前,戈那瑞林一般通过多肽化学合成的方法获得。
多肽化学合成技术已较为成熟[2,3],上海丽珠制药有限公司应用DCCI/tBoc固相化学合成技术,研制出了一条简单、有效的戈那瑞林合成工艺路线。但是,化学合成的多肽常常含有大量杂质,由于小部分杂质与目的产物在结构和理化性质上十分相近, 给多肽的分离纯化带来了困难。
反相液相色谱法在多肽的分离纯化中显示出优越的分离能力[4~12]。甘一如等[13]利用离子交换制备色谱和反相高效液相制备色谱在选择性上的差异成功地分离纯化出高质量的胸腺素α1,第一步采用阴离子交换色谱DEAE Sepharose Fast Flow除去粗品中的大部分非极性杂质,第二步用制备型反相高效液相色谱进一步纯化。最终胸腺素α1样品通过分析型反相液相色谱分析,其纯度达到95%以上。对于固相合成的戈那瑞林粗品,作者利用离子交换色谱和制备型反相高效液相色谱在选择性上的差异成功地分离纯化出高质量的戈那瑞林,其纯度达到98.5%以上,达到注射剂要求。同甘一如等[13]方法相比,本方法所制备的多肽纯度提高3%以上;同只用制备型反相高效液相色谱一步纯化相比,本方法上样量提高10倍。本方法有较高的经济效益。
1 实验
1.1 试剂、仪器和填料
1.1.1 试剂
乙腈、三氟乙酸(TFA)、醋酸钠、醋酸、氯化钠、三乙胺、磷酸,分析纯,上海化学试剂公司;注射用水、戈那瑞林粗品,自制;戈那瑞林标准品,国家药品生物制品检定所。
1.1.2 仪器
离子交换色谱系统,国产,自行组装;Waters Prep LC 4000 System、Waters 510 HPLC,Waters 公司。
1.1.3 填料
CM 25 Sepharose Fast Flow,Pharmacia公司。
CMC填料,国产。
制备型反相高效液相色谱填料:C18,300 mm×50 mm,15 μm,DeltaPakTM。
分析型反相高效液相色谱填料:C18,200 mm×4.6 mm,5 μm,DiamonsilTM。
1.2 步骤与条件
离子交换色谱:缓冲溶液A:50 mmol·L-1醋酸钠缓冲溶液,pH值6.0。缓冲溶液B:500 mmol·L-1氯化钠、50 mmol·L-1醋酸钠缓冲溶液,pH值6.0。流速为10 mL·min-1。用缓冲溶液A平衡柱子使pH值为6.0;用缓冲溶液A溶解粗品,用0.45 μm微孔滤膜过滤;上样,用缓冲溶液A平衡柱子使pH值为6.0;用缓冲溶液B洗脱,检测波长为280 nm,当紫外吸收值上升时收集样品,紫外吸收值不再上升时停止收集。

分析型反相高效液相色谱:以0.1 mol·L-1磷酸溶液(用三乙胺调节pH值至3.0)-乙腈(85∶15)为流动相,检测波长为220 nm,进样量为 20 μL。
1.3 方法
称取500 mg DCCI/tBoc化学合成的戈那瑞林粗品,用离子交换色谱缓冲溶液A溶解,定容至500 mL,得到1 mg·mL-1粗品溶液,经0.45 μm 微孔滤膜过滤后供离子交换色谱用。
离子交换后的样品不需要经过浓缩、脱盐处理以及调pH值,直接进行下一步制备型反相高效液相色谱操作。用制备型反相高效液相色谱的流动相A溶解离子交换后的样品,经0.45 μm微孔滤膜过滤后上样。
2 结果与讨论
2.1 紫外波长的确定
戈那瑞林含芳香族氨基酸酪氨酸和色氨酸,在紫外280 nm 处有强烈的吸收。而肽键的强吸收峰在紫外220 nm 处。为了有效地分离纯化,在离子交换色谱和制备型反相高效液相色谱纯化制备时,紫外波长设在280 nm处。为保证较高的检测灵敏度,在用分析型反相高效液相色谱检测样品时,将戈那瑞林的波长设在220 nm 处。在纯化制备和样品分析时分别采用不同的波长,这样可有效保证纯化样品的纯度和分析结果的可靠性。
2.2 化学合成戈那瑞林的反相高效液相色谱分析(图1)
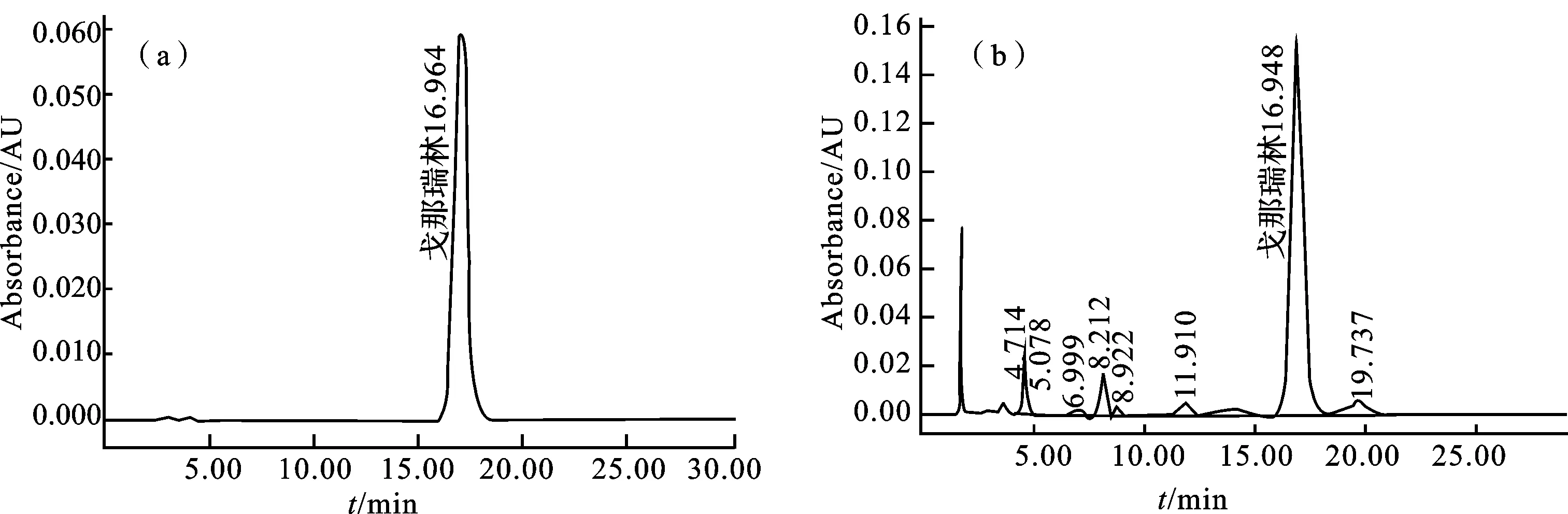
图1 戈那瑞林标准品(a)和粗品(b)的RP-HPLC图谱
由图1可知,戈那瑞林的保留时间为16.9 min,通过积分法测得戈那瑞林粗品的纯度为75.4%。化学合成法得到的戈那瑞林粗品组成较复杂,含有部分结构、性质和目标物相近的杂质,严重干扰了戈那瑞林的定性定量。在本条件下,目标峰和其周围的杂质峰能得到较好的分离,表明该条件适宜戈那瑞林粗品的分析,也适用于纯化戈那瑞林样品以及戈那瑞林纯品的分析。
2.3 离子交换色谱条件的选择
按 1.2方法用离子交换色谱对戈那瑞林进行CM 25 Sepharose Fast Flow离子交换分离,将离子交换后的戈那瑞林按1.2方法用分析型反相高效液相色谱分析,结果见图2。
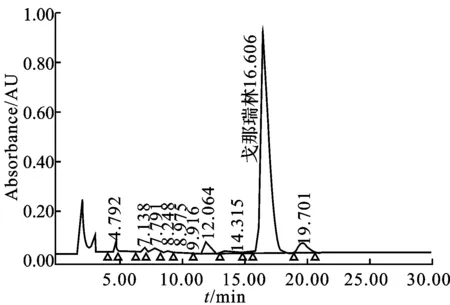
图2 戈那瑞林离子交换后的RP-HPLC图谱
由图2可见,选用CM 25 Sepharose Fast Flow填料离子交换后戈那瑞林和其它杂质得到一定的分离,主峰相对增高,杂峰相对降低。计算得戈那瑞林的纯度为85%、收率为80%,纯度有了较大提高。达到了对戈那瑞林粗品进行初步分离纯化的目的。更为重要的是,由于大部分非极性杂质不能被离子交换吸附,故可以通过离子交换法除去这部分非极性杂质。
实验还发现,按1.2方法用离子交换色谱对戈那瑞林进行CMC离子交换色谱,戈那瑞林也能被吸附,但杂质峰除去较少,基本达不到分离的目的。
同时考察了缓冲体系的 pH 值对分离效果的影响。选用 pH值为5.0、4.0的缓冲体系作进一步的研究,发现戈那瑞林也能被吸附,但杂质峰除去较少,基本达不到分离的目的。缓冲体系的 pH 值通过改变溶质的分配系数影响分离效果。选用pH值为6.0的缓冲体系可以达到分离的目的。
2.4 反相高效液相色谱条件的选择
对离子交换色谱所得的样品,按1.2方法用制备型反相高效液相色谱操作,上样没有流穿。反相高效液相制备色谱过程图谱见图3。所得戈那瑞林样品按1.2方法用分析型反相高效液相色谱分析,结果见图4。

图3 戈那瑞林反相高效液相色谱制备过程图谱
由图3可见,目标峰和杂质峰较好地分离,达到了很好的分离效果。

图4 纯化戈那瑞林反相高效液相图谱
由图4可见,经过反相高效液相色谱纯化几乎除去了所有杂质,戈那瑞林的纯度大大提高, 计算得戈那瑞林的纯度达到98.5%以上,收率达90%。
本实验曾尝试采用磷酸三乙胺体系进行反相高效液相色谱纯化,结果目标峰和杂质峰有一定程度重叠,不能分开,分辨率和收率都较低。因此,最终确定采用TFA体系进行反相高效液相色谱纯化。
2.5 反相高效液相色谱一步纯化戈那瑞林粗品的考察
本实验尝试采用反相高效液相色谱一步纯化戈那瑞林粗品。上样量为 500 mg,其它色谱条件与1.2相同。结果发现上样时有流穿现象,一步纯化后产物的纯度只达到89.9%;降低上样量到250 mg,上样时仍有流穿现象,一步纯化后产物的纯度只达到91.3%;进一步降低上样量到100 mg,上样时仍有流穿现象,一步纯化后产物的纯度只达到95.4%;最后上样量降低到50 mg,一步纯化后产物的纯度才达到98.8%。这可能是由于化学合成的戈那瑞林粗品中往往含有一些杂质,其分子结构与戈那瑞林十分相似,其反相色谱行为也与戈那瑞林十分接近,只用反相色谱法不能使化学合成的戈那瑞林得到有效的分离纯化。而这些杂质可以通过离子交换色谱有效地除去。
3 结论
针对DCCI/tBoc 固相化学合成戈那瑞林粗品,建立了一条联合应用离子交换色谱和反相高效液相色谱纯化路线。第一步采用阳离子交换色谱CM 25 Sepharose Fast Flow除去粗品中的大部分非极性杂质和部分极性杂质,第二步采用制备型反相高效液相色谱(C18色谱柱)除去大部分杂质。最终产品纯度达98.5%以上,纯化总收率达72%。本方法联合利用离子交换色谱和反相高效液相色谱各自的优势,大大提高了反相高效液相色谱的上样量和分离效率,有利于提高反相高效液相色谱的寿命和利用效率,从而获得较高的经济效益。离子交换后的戈那瑞林样品溶液不需要脱盐和浓缩处理以及调pH值,直接上样进行下一步的纯化,简化了操作,提高了产品的收率和质量。由于离子交换色谱和反相高效液相色谱都容易放大生产,故本方法易于放大生产。
参考文献:
[1] 田玲,路学智.瑞林类人工合成肽类药物的研究进展[J].中国新药杂志,2006,15(20):1723-1726.
[2] Verlander Michael.Industrial applications of solid-phase peptide synthesis.A status report[J].International Journal of Peptide Research and Therapeutics,2007,13(1-2):75-82.
[3] Uhmann Rainer,Radscheit Kurt.Process for the low-racemization preparation of peptide intermediates of the synthesis of gonadorelin and gonadorelin analogs,and new intermediates for this process[P].USP 4 691 008,1985-03-25.
[4] 韩香,顾军.高效液相色谱法在合成多肽分离与纯化中的应用[J].天津药学,2003,15(6):42-44.
[5] Andersson L,Blomberg L,Flegel M,et al.Large-scale synthesis of peptides[J].Biopolymers(Peptide Science),2000,55(3):227-250.
[6] 耿娟,王艳玲,陈丽颖,等.反相液相色谱在多肽及蛋白质分离分析中的应用[J].广州农业科学,2006,(12):133-135.
[7] 萨仁娜,毕力夫,苏秀兰.生物多肽的色谱分离[J].内蒙古医学院学报,2004,26(4):309-312.
[8] 黄鹤,万里鹏,吴蕾,等. 液相色谱法分离纯化固相合成的胸腺素α1[J].化学工业工程,2001,18(6):331-335.
[9] 张晓峰,陈庆森,庞广昌,等.液相色谱在多肽物质分离分析上的应用[J].食品科学,2006,27(3):239-241.
[10] Fields Cynthia G,Grab Beate,Lauer Janelle L,et al.Purification and analysis of synthetic,triple-helical"minicollagens" by revervsed-phase high-performance liquid chromatography[J].Analytic Biochemistry,1995,231(1):57-64.
[11] 高玲,余蓉.水蛭素12肽与瑞替普酶融合蛋白的反相色谱纯化及其一级结构确认[J].食品与药品,2008,10(1):8-12.
[12] Boysen Reinhard I,Hearn Milton T W.Purification of peptides from solid-phase peptide synthesis with RP-HPLC-Subscription Required Protocol[J].CSH Protocols;2006;doi:10.1101/pdb.prot4548.
[13] 甘一如,黄永东,吴蕾.应用液相色谱法分离纯化固相化学合成胸腺素α[J].化工学报,2004,55(3):497-500.
猜你喜欢
分析仪器(2022年5期)2022-10-14 09:58:04
中国石油大学学报(自然科学版)(2022年4期)2022-09-05 06:34:14
化工管理(2021年7期)2021-05-13 00:45:08
化学与粘合(2020年6期)2020-03-08 09:06:30
食品与机械(2018年5期)2018-07-14 03:15:24
科技视界(2017年25期)2017-12-11 20:30:32
橡胶工业(2016年2期)2016-02-23 21:36:51
医学美学美容·中旬刊(2015年2期)2015-10-21 19:58:27
河南科技(2014年15期)2014-02-27 14:12:29
无机盐工业(2013年1期)2013-03-19 23:25:40
