
2-苯氧乙胺盐酸盐的合成
2010-05-29 06:08方志文
武汉工程大学学报 2010年11期
方志文,陈 达,王 凯*,周 巍
(1.湖北省新型反应器与绿色化学工艺重点实验室,武汉工程大学化工与制药学院,湖北 武汉 430074;2.中国建筑第三工程局武汉中心医院药剂科,湖北 武汉 430070 )
0 引 言
2-苯氧乙胺盐酸盐(2-phenoxyethylamine,1)是制备热凝树脂、表面活性剂、矿石泡沫浮选剂和煤浮选调节剂的重要化工中间体[1],也是合成抗抑郁药奈法唑酮的关键医药中间体[2].2-苯氧乙胺的合成路线文献报道较多,但是产率不高,具体如图1所示.
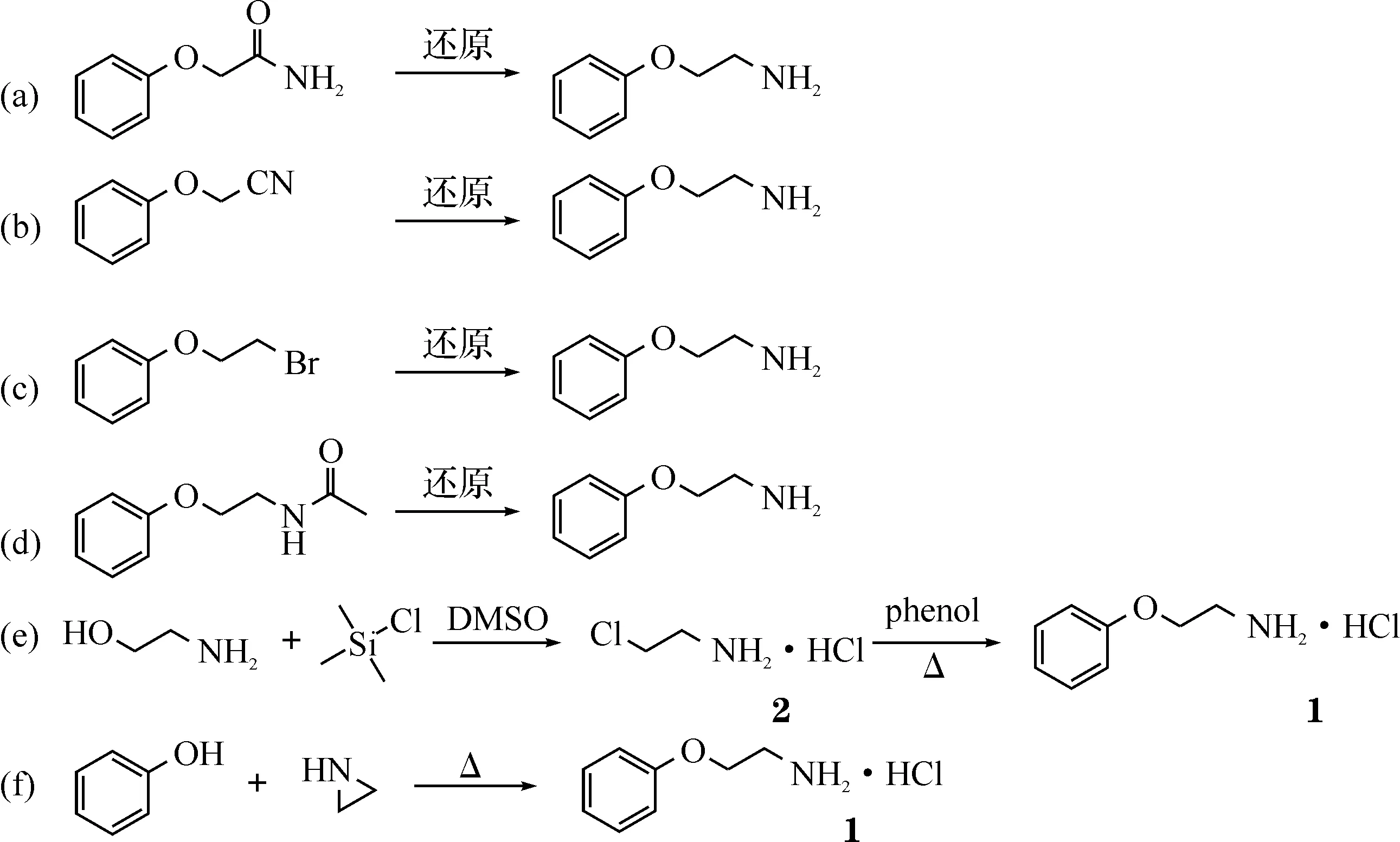
图1 2-苯氧乙胺的合成路线图解
路线a,b还原反应还原剂成本较高[3-4],且通常采用高压催化加氢,反应条件苛刻,不适宜工业化生产.路线c胺化易生成仲胺、叔胺等副产物[5],收率不高.路线d的原料难以制备[6],工业化成本较高.因此,本文以乙醇胺为原料,先与三甲基氯硅烷反应生成2-氯乙胺盐酸盐(2),再与苯酚缩合成功制得2-苯氧乙胺,成盐得到目标产物,总收率75%,如路线e所示.此外,本文在文献[7]的基础上,改进了路线f的操作条件,实现了对原料、溶剂进行回收套用,使反应收率从46.5%提高到57%.
1 实验部分
1.1 仪器与试剂
RY-1型熔点仪;varian Mercury-Vx 300型核磁共振仪(d6-DMSO为溶剂,TMS为内标);Finnigan Trace GC-MS型质谱仪.所用试剂均为分析纯.
1.2 合成部分
1.2.1 2-氯乙胺盐酸盐(2)的制备
向装有机械搅拌的250 mL三口烧瓶中加入乙醇胺24.4 g(0.40 mol)和三甲基氯硅烷86.9(0.80 mol).室温搅拌下,滴加二甲基亚砜7.8 g(0.1 mol),滴加完毕.继续搅拌反应30 min,随后通入HCl气体15 min.反应液过滤,滤饼用丙酮充分洗涤,干燥,得灰白色固体40.6 g,收率87.5%,m.p.143~148 ℃(文献值[8]: 146~148 ℃),未进一步纯化.
1.2.2 2-苯氧乙胺盐酸盐(1)的合成
a. 合成路线e:向500 mL的三口烧瓶中依次加入苯酚40 g(0.42 mol),蒸馏水300 mL,氢氧化钠33.68 g(0.84 mol),体系升温至80 ℃搅拌反应20 min.在反应液中分批加入2-氯乙胺盐酸盐(2)48.84 g(0.42 mol),继续搅拌反应2 h.反应液冷却至室温,用氯仿 (50 mL×4)萃取,合并有机相,用无水硫酸钠干燥,过滤,浓缩.粗品边搅拌边倒入2 mol/L氯化氢溶液20 mL中,大量白色固体析出,过滤,滤饼用石油醚充分洗涤,干燥,得白色固体得64 g,收率88%.
b. 合成路线f:在装有滴液漏斗和回流装置的1 L三口烧瓶中,加入苯酚95 g(1 mol),甲苯600 mL,加热回流,体系用气球密闭,缓慢滴入环乙胺10.75 g(0.25 mol)的甲苯溶液100 mL,回流反应,TLC检测反应进程.反应液冷却,用2 mol/L盐酸洗涤反应液 (200 mL×3),甲苯层用无水Na2SO4干燥(回收套用).酸层用NaOH溶液调至pH 10,再用氯仿萃取 (150 mL×4),氯仿层用无水硫酸钠干燥,浓缩,减压蒸馏得粗品30 g.馏分边搅拌边倒入2 mol/L氯化氢溶液10 mL中,大量白色固体析出,过滤,滤饼用石油醚充分洗涤,干燥,得白色固体25 g,产率57%.
2-苯氧乙胺盐酸盐的波谱数据:1H NMR(CDCl3)δ:1.83 (s,2H,NH2),3.0~3.2 (t,2H,CH2),3.9~4.1 (t,2H,CH2),6.8~7.0 (m,3H,ArH),7.2~7.4 (m,2H,ArH); MS (m/z):136 [M-1]+.
2 结果与讨论
对于合成路线e而言,在2-氯乙胺盐酸盐(2)的制备时,采用三甲基氯硅烷作为氯化试剂、二甲基亚砜作为催化剂,制备2-氯乙胺盐酸盐.此法相比传统的用二氯亚砜作为氯化试剂,反应条件温和,操作简便,且环境友好.同时,采用苯酚和2-氯乙胺亲核取代制备2-苯氧乙胺时,由于苯酚的亲核活性一般比伯胺强,所以反应生成的2-苯氧乙胺不会进一步与2-氯乙胺发生缩合反应生成多聚副产物.
对于合成路线f而言,制备2-苯氧乙胺时,在溶剂选择上,根据SN2反应特点和环乙胺的开环活性,改用沸点更高的非极性溶剂甲苯作为反应溶剂,而代替文献[7]采用的低沸点溶剂氯仿.在投料方式上,为减少多取代副产物的生成,采用在回流体系下,向母液缓慢滴加环乙胺的甲苯溶液的方法,能有效避免环丙胺的自身聚合.
此外,文献[7]采用减压蒸馏的方法用于分离2-苯氧乙胺和未反应的苯酚,但是苯酚的残留问题比较突出.因此,为了提高产品质量,本文在后处理中,均采用酸化成盐的方法,可以避免了苯酚残留问题.
本研究探索了一条以乙醇胺为原料,经氯代、缩合和成盐反应合成2-苯氧乙胺盐酸盐的新路线e,总收率75%.同时,结合反应物性质和反应原理,对合成路线f的操作条件进行优化,使反应收率从46.5%[7]提高到57%,提高了10.5%,并实现了对原料、溶剂进行回收套用,降低成本,所以,都具有潜在的工业化价值.
参考文献:
[1]Wei Y S,George P S.Prepation of phenoxyethenmaine [P]. US: 527612. 1994.
[2]Davis R,Whittington R,Bryson H M.Nefazodone,a review of its pharmacology and clinical efficacy in the management of major depression [J].Drugs,1997,53 (4): 608-636.
[3]Stirton A J. Amines [P].Swiss: 273953. 1951.
[4]董新荣,杨建奎.5-乙基-4- (2-苯氧乙基)-1,2,4-三唑-3-酮的合成[J].精细化工中间体,2003,33(3):23-25.
[5]Li Ai Jun,Zhou Xue Qin,Liu Dong Zhi. Improved synthesis of nefazodone from phenol [J]. Transactions of Tianjin University,2006,12(4):48-251.
[6]章小波,蒋永祥.抗抑郁药奈法唑酮中间体2-苯氧乙胺的合成工艺改进[J].精细化工中间体,2003,33(5):28-29.
[7]Ulrich H,Emanuel P,Karl F Z. The action of alcohols ethylenimines (aziridines) synthesis of β-amino ethers[J].Bar,1964,97(2):510-519.
[8]Wood,Thomas F. 1-Amino-2-haloethanes from 2-oxazolidones:US,2617825[P].1952-11-11.
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
广州化工(2022年7期)2022-04-26
汕头大学学报(自然科学版)(2020年4期)2020-12-14
有机氟工业(2019年2期)2019-08-12
天津药学(2016年5期)2017-01-16
化工生产与技术(2016年5期)2016-03-13
股市动态分析(2015年12期)2015-09-10
畜牧兽医学报(2015年3期)2015-07-05
应用化工(2014年1期)2014-08-16
无机化学学报(2014年12期)2014-02-28
