
PI3K/Akt/GSK-3β信号通路在肾小管上皮细胞缺血再灌注损伤中的调控作用及重组人红细胞生成素预保护效应*
2010-05-16 02:04周文祥杨永丽夏章晖聂祥智徐翠玲
华中科技大学学报(医学版) 2010年3期
周文祥, 杨永丽, 杨 晓△, 夏章晖, 韩 敏, 聂祥智, 徐翠玲
1武汉市第一医院肾内科,武汉 430022
2华中科技大学同济医学院附属协和医院肾内科,武汉 430022
3华中科技大学同济医学院附属梨园医院口腔科,武汉 430077
4华中科技大学同济医学院附属同济医院肾内科,武汉 430030
急性肾脏缺血再灌注(I/R)损伤是休克、严重创伤及肾移植过程中一种常见的临床病理生理现象,可导致急性肾衰竭(ARF)和移植肾功能丧失[1]。研究发现,在人和动物模型的肾脏缺血性损伤中,有大量肾小管上皮细胞凋亡和坏死[2],而减少肾小管上皮细胞的凋亡可减轻缺血肾脏组织的损伤[3],表明肾小管上皮细胞的凋亡在肾脏的缺血性损伤中起了重要作用。研究显示磷脂酰肌醇-3激酶/蛋白激酶B(PI3K/Akt)是一种重要的抗凋亡因子[4],它通过降低细胞凋亡的相关蛋白激酶Caspases家族、Bcl2等凋亡因子,在肾脏缺血再灌注损伤的动物模型中可以减少肾小管上皮细胞的凋亡从而发挥重要的保护作用。而糖原合成酶激酶3β(GSK-3β)作为 Akt的一个底物,在心脏[5]、脑[6]等脏器的缺血再灌注损伤细胞的凋亡过程中发挥了重要的作用,但其是否在肾脏缺血再灌注损伤细胞凋亡中发挥效应尚未见报道。最近实验发现重组人红细胞生成素(rHuEPO)在心脏[7]、肾脏[8]等脏器的缺血再灌注损伤中能够促进Akt的活性而减少细胞凋亡,从而发挥重要的器官保护作用[7]。本研究通过在肾小管上皮细胞(HK-2)缺血再灌注模型中利用特异性阻断剂分别阻滞Akt、GSK-3β活性,观察这些干预措施对HK-2细胞凋亡的影响,探讨PI3K/Akt/GSK-3β通路对HK-2缺血再灌注损伤过程中细胞凋亡的调控作用,另外,观察以rHuEPO预处理的HK-2缺血再灌注后细胞凋亡的变化,探讨rHuEPO预保护作用及可能机制。
1 材料与方法
1.1 主要试剂
抗霉素 A 、钙离子通道载体 A23187、LY294002、氯化锂(LiCl)、MTT(美国 Sigma),AnnexinⅤ-FITC凋亡试剂盒(南京凯基生物),兔抗人phospho-Akt Ser473多克隆抗体、兔抗人Akt多克隆抗体、兔抗人GSK-3β多克隆抗体、兔抗人phospho-GSK-3βSer9单克隆抗体(美国Cell Signaling Technology),兔抗人Caspase 3多克隆抗体(美国Santa Cruz),重组人促红细胞生成素(益比奥),沈阳三生公司惠赠。
1.2 细胞培养及实验设计
HK-2细胞株由同济医学院免疫学系实验室保存,以含10%胎牛血清的DMEM/F12培养液在37℃、5%CO2、饱和湿度条件下传代培养。正常培养的HK-2细胞同步化24 h后更换含10 μ mol/L抗霉素 A 及 1 μ mol/L A23187[9-10]的无血清DMEM/F12培养液孵育1 h,37℃预温的PBS清洗之后换成正常含血清培养液,以模拟体内细胞缺血再灌注损伤状态。实验分组如下:正常对照组、缺血再灌注(I/R)组、LY294002干预组、LiCl干预组、rHuEPO干预组、rHuEPO+LY294002双干预组、rHuEPO+LiCl双干预组,各干预组分别于缺血损伤前使用 LY294002(10 μ mol/L)、LiCl(20 μ mol/L)、rHuEPO(20 U/L)、rHuEPO+LY294002(20 U/L 、10 μ mol/L)、rHuEPO+LiCl(20 U/L 、20 μ mol/L)预孵育30 min,用抗霉素 A及 A23187诱导细胞缺血1 h后,更换为正常培养液继续培养2 h。
1.3 Western blot分析 Akt、GSK-3β和 Caspase 3 活性
以细胞裂解法提取蛋白,用BCA蛋白定量试剂盒测定蛋白浓度并配平,样本加入10%SDS-PAGE凝胶进行电泳,转膜至硝酸纤维素膜。一抗Akt Ser473(1∶1 000)、Akt(1 ∶1 000)、GSK-3β(1∶1 000)、GSK-3βSer9(1 ∶1 000)、Caspase 3(1 ∶1 000)和β-actin(1∶800)4℃孵育摇床过夜。二抗(1∶3 000)室温孵育2 h,ECL化学发光试剂盒显色、曝光、显影、定影。用UVP凝胶成像扫描系统对胶片条带行定量吸光度(A)测定。
1.4 流式细胞仪检测细胞凋亡
胰酶消化细胞,PBS清洗2次,将细胞重悬于300 μ L 结合缓冲液,分别加入 5 μ L Annexin Ⅴ试剂和PI混匀,室温避光孵育15 min后上机。
1.5 MTT法检测细胞存活率
细胞以每孔4 000个接种于96孔板,常规培养、同步。各分组给予干预因素。分别加入MTT(终质量浓度5 g/L),37℃孵育4 h,弃上清,加入二甲基亚砜150 μ L,室温溶解 10 min,分光光度计 570 nm 波长读取A值。每组设6个复孔,实验重复3次。
1.6 统计学处理
2 结果
2.1 缺血再灌注及阻断剂LY294002、LiCl对 HK-2细胞凋亡率、存活率的影响
2.1.1 流式细胞仪检测结果 正常对照组[(3.3±0.8)%]和I/R组[(15.2±1.4)%]细胞凋亡率差异有统计学意义(P<0.01),经阻断剂 LY294002预处理后缺血再灌注细胞的凋亡率上调为[(18.2±2.1)%],经阻断剂 LiCl预孵育后缺血再灌注细胞的凋亡率达到[(12.3%±0.8)%],与正常对照组和I/R组相比,差异均有统计学意义(均P<0.05),见图1(A~D)。
2.1.2 M TT检测结果 正常对照组(0.824±0.016)和I/R组(0.313±0.022)细胞 A值差异有统计学意义(P<0.05),经阻断剂 LY294002预处理后缺血再灌注细胞的 A值下调为(0.201±0.026),经阻断剂 LiCl预孵育后缺血再灌注细胞的A值达到(0.468±0.030),与正常对照组和I/R组相比,差异均有统计学意义(均P<0.05),见图2(A~D)。
2.2 rHuEPO预孵育对HK-2细胞缺血再灌注细胞凋亡率、存活率的影响
2.2.1 流式细胞仪检测结果 与I/R组[(15.2±1.4)%]相比,rHuEPO干预组[(11.1±1.6)%]细胞凋亡率明显降低(P<0.05);rHuEPO+LY294002双干预组细胞凋亡率(13.4±1.9)%、rHuEPO+LiCl双干预组细胞凋亡率(7.5±1.3)%,与rHuEPO干预组相比,差异均有统计学意义(均 P<0.05),见图1(E~G)。
2.2.2 MT T检测结果 与I/R组(0.313±0.022)相比,rHuEPO干预组(0.574±0.016)A值明显升高(P<0.05);rHuEPO+LY294002双干预组细胞A值(0.424±0.015)、rHuEPO+LiCl双干预组细胞 A值(0.674±0.011)与rHuEPO干预组相比,差异均有统计学意义(均P<0.05),见图2(E~G)。

图1 流式细胞仪检测各组HK-2细胞凋亡率Fig.1 Apoptotic ratio of HK-2 cells in each g roup examined by flow cytometry

图2 不同干预措施对HK-2细胞存活力的影响(MT T法)Fig.2 Viability of HK-2 cells in each group measured by M TT
2.3 缺血再灌注对HK-2细胞Akt、GSK-3β和Caspase 3酶活性的影响
Western blot分析结果显示:与正常对照组相比,再灌注0、0.5 h出现一过性的Akt活性水平上调和GSK-3β活性水平下调,而再灌注1 h出现Akt活性水平下降和GSK-3β活性水平上升,再灌注2、4 h Akt活性明显低于正常对照组,而GSK-3β活性明显高于正常对照组(P<0.05或P<0.01),见图3。
与正常对照组比较,再灌注0、0.5 h时均未见Caspase 3活性变化,而再灌注1 h时出现Caspase 3活性的升高,再灌注 2、4 h时 Caspase 3活性均明显高于正常对照组(均P<0.01),见图3。
2.4 阻断剂 LY294002、LiCl对 HK-2 细胞 Akt、GSK-3β 和Caspase 3活性的影响
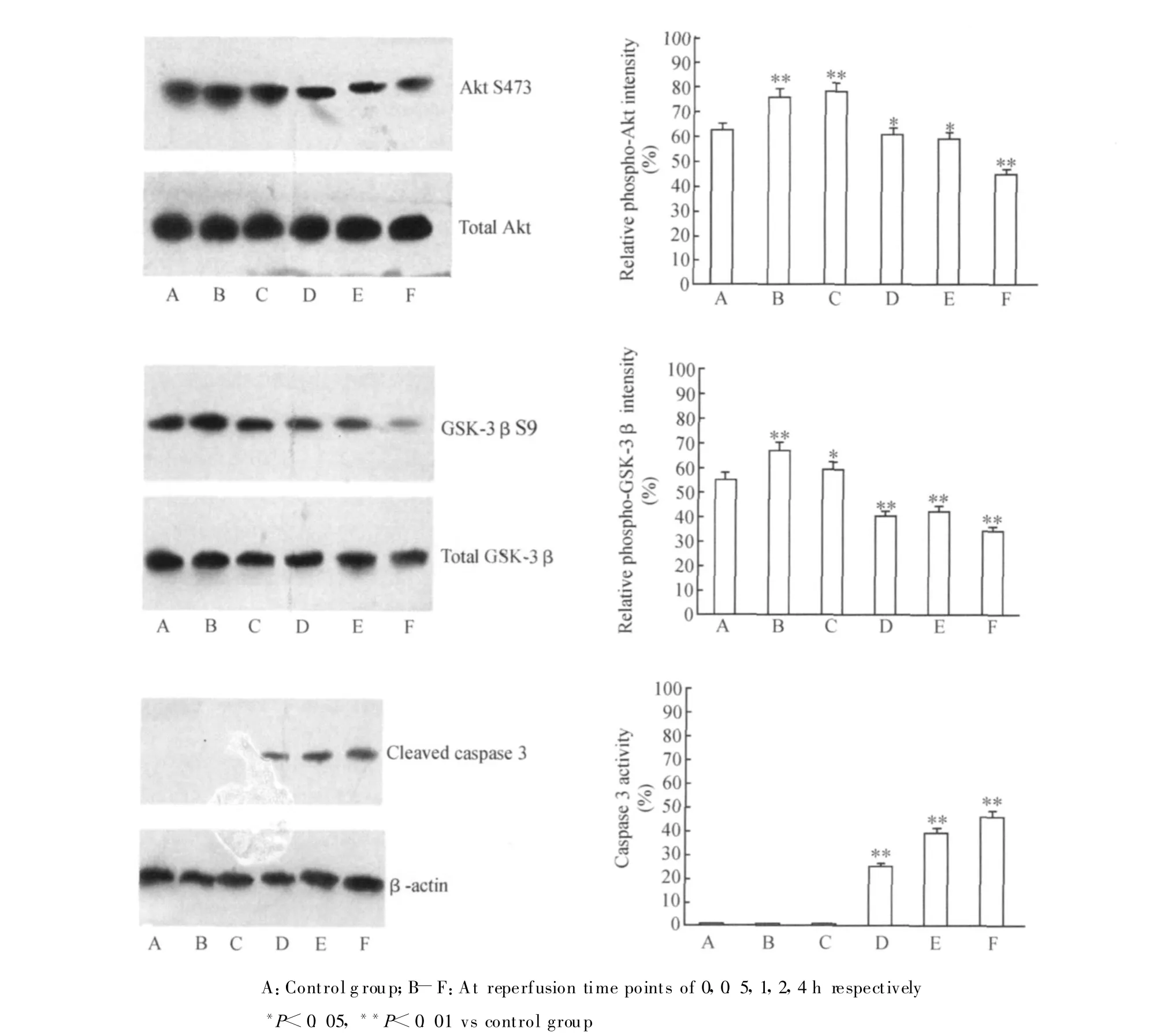
图3 缺血再灌注对HK-2细胞内Akt、GSK-3β及Caspase 3活性的影响Fig.3 Effects of I/R on activities of Akt,GSK-3β and Caspase 3 in HK-2 cells
与I/R组相比,阻断剂LY294002干预后,细胞内Akt活性水平降低,GSK-3β活性升高;阻断剂LiCl干预后,细胞内Akt活性水平升高,GSK-3β活性降低。I/R组HK-2细胞内Caspase 3活性水平较正常对照组显著上调(P<0.05);阻断剂LY294002干预后HK-2细胞内Caspase 3活性较I/R组显著增高(P<0.05);而阻断剂 LiCl干预组HK-2细胞内Caspase 3活性水平较I/R组明显下降(P<0.05),见图4。
2.5 rHuEPO预孵育对Akt、GSK-3β和Caspase 3活性的影响
与I/R组相比,rHuEPO干预组HK-2细胞内Akt活性水平显著上调、GSK-3β活性水平显著下调(均P<0.05);与rHuEPO干预组相比,rHuEPO+LY294002双干预组细胞内Akt活性水平下调、GSK-3β活性水平上调,而 rHuEPO+LiCl双干预组细胞内Akt活性表达上调 、GSK-3β活性水平下调(均P<0.05),见图4。
rHuEPO干预组与I/R组相比,Caspase 3活性水平显著下降(P<0.05)。与 rHuEPO组相比,rHuEPO+LY294002双干预组细胞内Caspase 3活性水平升高、rHuEPO+LiCl双干预组细胞内Caspase 3活性水平降低,差异均有统计学意义(均P<0.05),见图4。
3 讨论
磷脂酰肌醇-3激酶/蛋白激酶B(PI3K/Akt)通路是介导细胞存活的一条经典通路[7],活化的PI3K通过3-磷酰化磷酸肌醇酯和磷酸肌醇依赖性激酶(PDK)共同作用而激活Akt,Akt的激活是通过其氨基酸残基位点Ser473和Thr308的磷酸化实现的。研究表明PI3K/Akt在心脏[7,11]、肾脏[8]等缺血再灌注损伤中发挥了重要的保护作用。

图4 不同组别HK-2细胞内Akt、GSK-3β及Caspase 3相对活性Fig.4 T he relative activities of Akt,GSK-3β and Caspase 3 in each g roup of HK-2 cells
糖原合成酶激酶 3β[12](glycogen synthase kinase-3p,GSK-3β)是Akt重要的下游因子之一[12],GSK-3β广泛地表达于机体的各种组织和细胞中,其肽链结构中富含丝氨酸/苏氨酸残基。Tyr216残基被磷酸化后其活性被激活,而Ser9残基被磷酸化后其活性被抑制。最新发现GSK-3β在多种信号传导过程中起关键作用,它不仅通过激活糖原合成酶调节细胞糖代谢从而调控细胞能量代谢过程,更重要的是在细胞的生长、分化、突变和细胞凋亡等生命活动中也具有重要的调控作用。研究表明,在多种正常组织细胞以及多数类型的肿瘤细胞中,GSK-3β的激活可以进一步激活细胞凋亡的相关蛋白激酶,如BAX[13],c-jun[14],Caspases家族[15]等,从而诱导细胞凋亡。另有研究发现,在心肌[11,16],脑[6]等缺血再灌注中GSK-3β的磷酸化水平降低,活性增强,促进了缺血再灌注损伤脏器中细胞的凋亡。在肾脏也发现有GSK-3β的表达,其在慢性肾炎的发展中起到了重要作用[15]。但GSK-3β是否在肾脏缺血再灌注损伤肾小管上皮细胞的凋亡中起作用尚未见报道。
Caspase的活化是凋亡发展过程中的关键环节,Caspase级联效应一旦激活,意味着凋亡的发生不可避免。近年来研究多表明在心肌的缺血性损伤中激活 Caspase 3的同时也发现有 GSK-3β的激活[11],当GSK-3β上游的信号转导分子受到抑制导致GSK-3β激活时,就会促进 Caspase 3的激活,从而造成细胞凋亡。
本实验用抗霉素A及A23187[9-10]制作 HK-2细胞的缺血再灌注损伤模型,模拟体内缺血再灌注状态。用 PI3K/Akt阻断剂 LY294002、GSK-3β阻断剂 LiCl分别阻断 Akt、GSK-3β活性,以观察Akt、GSK-3β是否在HK-2的缺血再灌注损伤中发挥作用,并观察PI3K/Akt是否通过GSK-3β起作用。在实验中,首先观察到缺血再灌注损伤诱导HK-2细胞凋亡率增加并细胞活力下降,细胞内Akt(Ser473)及GSK-3β(Ser9)水平一过性升高后也呈时间依赖性下调,Caspase 3活性呈时间依赖性上调,表明HK-2细胞缺血再灌注过程中Akt活性下调而 GSK-3β活性升高。使用PI3K/Akt阻断剂LY294002阻滞Akt活性后,细胞内Caspase 3活性进一步上调且细胞凋亡率升高,说明Akt在减少缺血再灌注细胞凋亡过程中发挥保护作用。研究发现Akt活性阻滞的同时伴有GSK-3β(Ser9)表达水平进一步下调,说明Akt活性的抑制可促进GSK-3β活性的上调。当使用 GSK-3β阻断剂 LiCl阻滞GSK-3β活性后,Caspase 3活性下调,细胞凋亡率下降,说明GSK-3β活性的上调在缺血再灌注损伤HK-2细胞凋亡中发挥重要作用。因此推论,正常细胞内Akt活性的存在,使其下游因子GSK-3β活性水平受到抑制,保护细胞免于凋亡;而缺血再灌注损伤抑制了Akt活性,从而降低了其对下游因子GSK-3β蛋白的磷酸化作用,GSK-3β活性的升高参与了缺血再灌注损伤诱导肾小管上皮细胞的凋亡。
最近研究显示rHuEPO预孵育对心脏[7]、肾脏等[8]脏器的缺血再灌注损伤有保护作用,并且揭示了部分的作用机制。Sharples等[8]研究发现rHuEPO可使Akt(Ser473)表达水平上调,降低Caspase 3,8,9活性,促进Bcl-XL表达,从而减少细胞的凋亡。本实验研究发现rHuEPO干预可使细胞凋亡率下降、细胞活力提高,同时伴有 Akt(Ser473)、GSK-3β(Ser9)水平上调,Caspase 3活性降低。较单纯rHuEPO干预相比,rHuEPO+LY294002双干预后,细胞凋亡率升高,GSK-3β活性水平提高,Caspase 3酶活性上调;rHuEPO+LiCl双干预后,细胞凋亡率下降,Caspase 3酶活性下调,由此推论,rHuEPO通过促使上调Akt活性,抑制其下游因子GSK-3β的活性,从而减轻了缺血再灌注损伤所致的细胞凋亡。
综上所述,缺血再灌注损伤抑制了HK-2细胞的Akt信号传导通路,从而激活了其下游因子GSK-3β、Caspase 3的活性,可能是其导致HK-2细胞凋亡的信号转导通路之一。rHuEPO能够提高Akt活性水平,抑制GSK-3β活性,使HK-2在缺血再灌注损伤中形成了耐受,为其成为潜在治疗缺血性损伤的药物提供了实验依据。GSK-3β的激活可以诱导 HK-2的凋亡,但其确切的下游机制有待进一步探讨。
[1] Kelly K J,M olitoris B A.Acute renal failure in the new millennium:time to consider combination therapy[J].Semin Nephrol,2000,20(1):4-19.
[2] Padanilam B J.Cell death induced by acute renal injury:a perspective on the contributions of apoptosis and necrosis[J].Am J Physiol Renal Phy siol,2003,284(4):F608-F627.
[3] De Vries B,Matthijsen R A,Van Bijnen A A,et al.Ly sophosphatidic acid prevents renal ischemia-reperfusion injury by inhibition of apoptosis and complement activation[J].Am J Pathol,2003,163(1):47-56.
[4] Datta S R,Brunet A,Greenberg M E.Cellular survival:a play in three Akts[J].Genes Dev,1999,13(22):2905-2927.
[5] Yin H,Chao L,Chao J.Adrenomedullin protects against myocardial apoptosis after ischemia/reperfusion through activation of Akt-GSK signaling[J].Hy pertension,2004,43(1):109-116.
[6] Linseman D A,Butts B D,Precht T A,et al.Gly cogen synthase kinase-3 phosphorylates bax and promotes its mitochondrial localization during neuronal apoptosis[J].J Neurosci,2004,24(44):9993-10002.
[7] Cai Z,Semenza G L.Phosphatidylinositol-3-Kinase signaling is required for ery thropoietin-mediated acute protection against myocardial ischemia/reperfusion injury[J].Circulation,2004,109(17):2050-2053.
[8] Sharples E J,Patel N,Brown P,et al.Erythropoietin protects the kidney against the injury and dy sfunction caused by ischemia-reperfusion[J].J Am Soc Nephrol,2004,15(8):2115-2124.
[9] Xie J,Guo Q.Apoptosis antagonizing transcription factor protects renal tubule cells against oxidative damage and apoptosis induced by ischemia-reperfusion[J].J Am Soc Nephrol,2006,17(12):3336-3346.
[10] Nilakantan V,Liang H,Maenpaa C J,et al.Differential patterns of peroxy nitrite mediated apoptosis in proximal tubular epithelial cells following ATP depletion recovery[J].Apoptosis,2008,13(5):621-633.
[11] Song J Q,T eng X,Cai Y,et al.Activation of A kt/GSK-3beta signaling pathway is involved in intermedin(1-53)protection against myocardial apoptosis induced by ischemia/reperfusion[J].Apoptosis,2009,14(9):1061-1069.
[12] Hardt S E,Sadoshima J.Glycogen synthase kinase-3β a novel regulator of cardiac hy pertrophy and development[J].Circ Res,2002,90(10):1055-1063.
[13] Somervaille T C,Linch D C,Khwaja A.Growth factor withdrawal from primary human ery throid progenito rs induces apoptosis through a pathway involving glycogen sy nthase kinase-3 and Bax[J].Blood,2001,98(5):1374-1381.
[14] Choi Y K,Kim Y S,Choi I Y,et al.25-hydroxycholesterol induces mitochondria-dependent apoptosis via activation of glycogen synthase kinase-3beta in PC12 cells[J].Free Radic Res,2008,42(6):544-553.
[15] Gong R,Rifai A,Dworkin L D.Activation of PI3K Akt GSK3b pathway mediates hepatocy t growth facto r inhibition of RANTES expression in renal tubular epithelial cells[J].Biochem Biophys Res Commun,2005,330(1):27-33.
[16] Gao H K,Yin Z,Zhou N,et al.Glycogen synthase kinase 3 inhibition protects the heart from acute ischemia-reperfusion injury via inhibition of inflammation and apoptosis[J].J Cardiovasc Pharmacol,2008,52(3):286-292.
猜你喜欢
四川农业科技(2018年6期)2018-03-17
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
成都医学院学报(2016年4期)2016-03-26
中外医疗(2015年11期)2016-01-04
华南农业大学学报(2015年5期)2015-12-04
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
西南军医(2015年6期)2015-01-23
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28

- 华中科技大学学报(医学版)的其它文章
- 868例感染性心内膜炎感染部位与病原菌分析