
Lipids-induced Apoptosis Is Aggravated by Acyl-coenzyme A: Cholesterol Acyltransferase Inhibitor△
2010-04-20 01:34JianlingTaoXiongzhongRuanHangLiXuemeiLiandXuewangLi
Chinese Medical Sciences Journal 2010年2期
Jian-ling Tao, Xiong-zhong Ruan, Hang Li, Xue-mei Li, and Xue-wang Li
1Department of Nephrology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100730, China
2Centre for Nephrology, University College London Medical School, Royal Free Campus, London, NW3 2PF, UK
THE concept of “glomerular atherosclerosis” was proposed in 1982,1suggesting that atherosclerosis and glomerulosclerosis share common pathogenic mechanisms.2Cardiovascular complications caused by accelerated atherosclerosis constitute the largest single cause of mortality in chronic renal failure patients.3Atherosclerotic plaques develop as a consequence of the accumulation of circulating lipids and the subsequent migration of inflammatory cells (macrophages and T cells) and vascular smooth muscle cells (VSMCs).
Apoptosis has been found in atherosclerosis and is increasingly observed in plaque development.4,5Both macrophages and VSMCs, the two major cell types involved, bear markers of apoptosis.6Apoptosis of VSMCs may contribute to the accumulation of plaque.7,8Apoptotic macrophages were detected in the lipid-rich core of atherosclerotic lesions.9Macrophage apoptosis is significantly higher at sites of plaque rupture and thrombosis in patients with sudden coronary death.10
The uptake of modified lipoproteins mainly by macrophages leads to the accumulation of cholesterol esters (CE) and the formation of macrophage-derived foam cell, the hallmark of fatty streak.11As the lesion progressed, free cholesterol (FC) accumulated and resulted in apoptosis.12,13
The enzyme responsible for cholesterol esterification is acyl-coenzyme A: cholesterol acyltransferase 1 (ACAT1) located in endoplasmic reticulum (ER) in cells.14Chang et al15pointed out that the main role of ACAT in intracellular cholesterol homeostasis is to guard against excessive buildup of cholesterol in ER. It would be advantageous to maintain the cholesterol concentration in ER low enough so that subtle changes in this parameter could produce sensitive signals for regulating sterol-regulated enzymes or proteins located in ER.16
When an ACAT inhibitor (ACATI) 58-035 was added to reduce the amount of CE, significant toxicity and cell death were observed.17It was shown that cholesterol loading in macrophages with ACATI added could induce accumulation of FC in ER and lead to apoptosis mediated by ER stress pathway.18,19
In this study, we observed the apoptosis of the cells involved in atherogenesis induced by oxysterol or modified lipoprotein, and the effect of ACATI on apoptosis in the context of intracellular cholesterol homeostasis change.
MATERIALS AND METHODS
Low density lipoprotein (LDL) isolation and acetylated LDL (AcLDL) preparation
Human LDL was isolated from fresh human plasma by sequential ultracentrifugation.20The degree of lipid peroxidation of the LDL was estimated as the concentration of thiobarbituric acid reactive substances (TBARS)21and the result was expressed as nmol malondialdehyde (MDA)/mg LDL. The levels of TBARS in native LDL used in this study were lower than 0.1 nmol MDA/mg LDL. AcLDL was prepared by reaction of LDL with acetic anhydride.22
THP-1 cells culture
THP-1 monocytes (American Type Culture Collection, Manassas, VA, USA) were maintained at 37°C in humidified 5% CO2atmosphere, in suspension at a cell density of 2.0×105-8.0×105/mL in RPMI 1640 containing 10% fetal bovine serum (FBS) (v/v), 2 mmol/L glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.05 mmol/L 2-mercaptoethanol (Sigma, Poole, UK). The cells were then cultured in phorbol 12-myristate 13-acetate (PMA) (Sigma) at a final concentration of 160 nmol/L for five days until complete differentiation into macrophage-like cells. All reagents for cell culture were obtained from GIBCO BRL (Paisley, UK) unless stated otherwise.
Human vascular smooth muscle cells (VSMCs) culture
Human aortic smooth muscle cells (TCS Cellworks, Liverpool, UK) were grown in TCS smooth muscle growth medium supplemented with 5% FBS. The cells were incubated at 37°C in humidified 5% CO2atmosphere. Medium was changed every 2-3 days, and passages 3-6 were used for experiments.
Protein determination and cell proliferation and cy- totoxicity assay
Protein concentration was determined by modified Lowry assay.23,24Human VSMCs were plated in 96-well plates (Falcon, Oxford, UK) at a density of 8 000 cells/well in standard culture medium (TCS Cellworks) until confluent. The cells were synchronized to the quiescent state by incubation in Dulbecco's modified Eagle's medium (DMEM) (GIBCO) containing 0.2% bovine serum albumin (BSA) (essentially fatty acid free) (Sigma) for 24 hours, then each well was washed with phosphate buffered saline (PBS) intensively. The cells were incubated for 24 hours in five different types of medium: DMEM containing 5% lipoprotein-deficient serum (LPDS) (GIBCO) as control, 1-15 μg/mL of 25-hydroxycholesterol (25OHC), 10-75 μg/mL of cholesterol, 25-200 μg/mL of LDL, and 25-100 μg/mL of AcLDL. For proliferation assay, the cells were then labelled for 18 hours with 1 μCi [3H] thymidine (Amersham, Buc- kinghamshire, UK), washed with PBS, trypsinized for 5-10 minutes, and harvested. Radioactivity associated with replicative DNA synthesis of proliferating cells was measured with a scintillation counter. In the [3H] incorporation experiment, the proliferation was represented by counts per minute (CPM). In toxicity assay, we detected lactate dehydrogenase (LDH) release in the supernatant and LDH in lysed cells with an enzyme assay following the instructions from In Vitro Toxicity Assay Kit Lactate Dehydrogenase (Sigma, Saint Louis, MS, USA). The cytotoxicity was represented by the ratio of LDH detected in the supernatant (assumed to be released from the dead cells) to the total amount of LDH detected. The sum was assumed as the total cell number, which was the final normalization index.
4’,6-diamidino-2-phenylindole (DAPI) staining
Human VSMCs were plated into eight-well plates (Nunc, Naperville, IL, USA) at 1×104cells/well. After reaching 60%-70% confluence, the cells were washed with DMEM and the culture medium were replaced by DMEM containing 0.2% BSA (as control), 200 nmol/L of ACATI FR 179254 (Calbiochem, Nottingham, UK), 15 μg/mL of 25OHC, or 200 nmol/L of ACATI FR179254 plus 15 μg/mL of 25OHC for 24-hour incubation. THP-1 cells were plated into eight- well plates at 8×104cells/well until fully differentiated by PMA for five days. The differentiated THP-1 cells were washed with serum-free RPMI and then cultured for 24 hours in four types of medium: serum-free RPMI medium (as control), 50 μg/mL of AcLDL, 100 μg/mL of AcLDL, and 50 μg/mL of AcLDL plus 800 nmol/L of ACATI FR 179254.
The treated cells were washed once with 1 μg/mL of DAPI (Roche Diagnostics GmbH, Mannheim, Germany) dissolved in methanol, then incubated with the stain for 15 minutes at 37°C in dark, and washed again with methanol. The plates were analyzed by fluorescence microscopy with 340/380 nm excitation filter and LP (long-pass) 430 nm barrier filter. Digital images were obtained with a C-4000 ZOOM Olympus digital camera (Olympus America, Inc.). The percentage of apoptotic cells was calculated based on the results of cell count in five different visual fields, the total number of cells counted in each field being more than 500.
Annexin-V and propidium iodide (PI) staining
Human VSMCs were seeded into 12-well plates (Nunc Inc., Naperville, IL, USA) at a density of 7×104cells/well. The following treatment prior to harvest was the same as in DAPI staining. The cell pellets were washed with PBS twice, then stained with Annexin-V-FLUOS (Roche Applied Science, Nonnenwald, Penzberg, Germany) labeling solution following the manufacturer's instructions. Stained cells were immediately analyzed by fluorescence-activated cell sorter (FACS) with a flow cytometer EPICS XL-MCL (Coulter, CA, USA).
Caspase-3, -7 activities and cell viability assay
THP-1 cells were seeded into opaque-walled 96-well plates (Nunc, Naperville, IL, USA) at a density of 2×104cells/well in 100 μL of RPMI 1640 medium. After fully differentiated, the cells were washed with RPMI 1640 medium twice, and cultured for 24 hours in three different media: 100 μL of RPMI 1640 medium (as control), 100 μg/mL of AcLDL, and 100 μg/mL of AcLDL plus 10 μg/mL of ACATI 58-035 (3-[decyldimethylsilyl]-N-[2-(4-methylphenyl)-1-phenylet- hyl] propanamide, kindly donated by Novartis, Basel, Switzerland). Caspase-3, -7 activities and viability of the cells were studied according to the protocols provided by the manufacturer of CellTiter-Glo®Reagent and Caspase-Glo®3/7 Reagent (Promega, Madison, WI, USA). Briefly, 100 μL of each of these two reagents were added into two sets of identically treated wells. The wells containing only experimental medium without cells were set as background. The mixtures were incubated at room temperature for 10-30 minutes. Measure the luminescence of each set of wells with a plate-reading luminometer, following the instructions provided by the manufacturer. The final reading was defined by subtracting the average reading detected from the background wells. The apoptosis was represented by the relative caspase activities after normalization by cell viability.
Intracellular FC and CE assay
THP-1 cells were seeded into 12-well plates at a density of 4×105in 2 mL of culture medium until fully differentiated. The cells were cultured for 24 hours in serum-free RPMI 1640 (as control), 100 μg/mL of AcLDL, or 100 μg/mL of AcLDL plus 10 μg/mL of ACATI 58-035. Modified method for determination of intracellular FC and total cholesterol (TC) was applied.25,26Briefly, re-suspend the cell pellet in 1 mL of chloroform/methanol 2∶1 (v/v); leave the pellet dry after sonification and dissolve it in 10 N of NaOH for protein assay. Remove the liquid phase into clean glass tubes. After chloroform was evaporated in vacuum, heat the tubes for 20 minutes at 105°C in an oven. Re-suspend the samples in 95% ethanol. Cholesterol and cholesteryl oleate were prepared in chloroform in stock at a concentration of 10 μg/mL and the standard solutions containing 0-300 ng cholesterol or 0-600 ng cholesteryl oleate were successively diluted in 95% ethanol. For the compositions of solutions used for determination of FC and TC, we resorted to references 25 and 26. Take aliquots of samples for FC and TC assays, and add prepared assay solutions. After incubation for 30 minutes at 37°C, fluorescence was measured with a spectrophotometer (excitation, 325 nm; emission, 415 nm). The actual amounts of FC and TC were obtained from the standard curves, and normalized by protein mass. The CE amount was determined by subtracting FC from TC. All the chemicals and organic solvents used in this assay were produced by Sigma.
Statistical analysis
All results in this study were expressed as the relative ratio over control (as 100%) unless otherwise indicated. Within an experiment, duplicate, triplicate, or quadruplicate wells were used for each condition or treatment. Data were collected from at least three separate experiments and presented as means±SD. SPSS11.0 software was used to analyze difference between samples with one-way analysis of variance (ANOVA), and S-N-K post-test for difference between selected pairs of samples. P<0.05 was considered statistically significant.
RESULTS
Cytotoxicity and effects of lipids on the proliferation of VSMCs
In our assay, 25OHC had biphasic effects on VSMC proliferation: at low concentration (below 5 μg/mL), it stimulated cell proliferation, but turned to inhibition as concentration reached 15 μg/mL. Meanwhile, in LDH release assay, the cytotoxicity of 25OHC appeared at the concentration of 5 μg/mL and intensified as concentration increased up to 15 μg/mL. In the concentration range studied (10-75 μg/mL), cholesterol in vitro inhibited VSMC proliferation only at concentrations above 50 μg/mL, and exerted cyto- toxicity only at the concentration of 75 μg/mL. LDL at the concentration of 25-200 μg/mL demonstrated dose-dependent stimulation on VSMC proliferation as the [3H] incorporation increased significantly compared with control, but no toxic effects were observed. AcLDL exhibited dose- dependent inhibition on cell proliferation in the studied range of 25-100 μg/mL, and showed cytotoxicity at the concentration ranging from 25 to 100 μg/mL (Fig. 1).
25OHC-induced apoptosis in VSMCs and the additive effect of ACATI on apoptosis
Since 15 μg/mL of 25OHC inhibited VSMC proliferation significantly, we investigated whether there existed apoptosis, and what the effect of ACATI was on this phenomenon. Three methods were used for detecting apoptosis, including Annexin-V and PI staining, DAPI staining, and caspase-3, -7 assay.
Compared with control, 200 nmol/L of ACATI FR 179254 alone could not induce apoptosis after incubation in DMEM medium containing 2% LPDS for 24 hours, as the percentage of Annexin-V positive cells detected by FACS did not increase significantly. 25OHC at 15 μg/mL significantly induced VSMC apoptosis as the ratio of apoptotic cell percentage between treated cells over control increased to 248.79%±12.51%, which was further enhanced by 200 nmol/L of FR 179254 to 293.97%±44.93% over control. The results from DAPI staining conformed with those from Annexin-V staining. After treatment with 25OHC, the apoptotic cells with condensed nuclei became remarkably visible. In cells treated with both ACATI FR179254 and 25OHC, more apoptotic cells were observed than in those treated with 25OHC alone (Fig. 2).
AcLDL-induced apoptosis in PMA differentiated THP-1 cells and the additive effect of ACATI on apoptosis
Macrophage is the main type of cell involved in the pathogenesis of atherosclerosis as it uptakes modified lipids including AcLDL and oxidized LDL (oxLDL) via scavenge receptors, the expression of which is not regulated by intracellular cholesterol contents. Therefore, we studied if AcLDL could induce apoptosis in PMA differentiated THP-1 cells and further observed if ACATI could augment the apoptosis. In terms of the percentage of apoptotic cells, more DAPI stained cells were observed in 50 μg/mL AcLDL treated group than in control (5.60%±1.68% vs. 1.90%± 0.14%, P<0.05). With the concentration of AcLDL doubled, the apoptotic cell percentage increased to 24.57%±3.75%, significantly higher than that with 50 μg/mL AcLDL (P< 0.001). Adding 800 nmol/L of FR 179254 significantly increased the apoptotic cell percentage compared with 50 μg/mL AcLDL treatment (11.93%±3.75% vs. 5.60%± 1.68%, P<0.05) (Fig. 3).
The above data led us to expect an increase in caspase-3, -7 activity when macrophages were lipid-loaded and a further increase when another ACATI, 58-035, was tested. As revealed in observation, 100 μg/mL of AcLDL significantly increased the caspase-3, -7 activity to 246.67%± 39.65% over control (P<0.01 compared with control), and 10 μg/mL of ACATI 58-035 increased the caspase-3, -7 activity to 330.91%±68.29% (P<0.05 compared with AcLDL treated cells), which showed that ACATI augmented the effect of AcLDL.
Augmentative effect of ACATI on apoptosis possibly caused by FC accumulation
By measuring the content of intracellular FC and CE, we tried to find out if intracellular cholesterol homeostasis was changed in THP-1 cells in apoptosis. THP-1 cells cultured in 100 μg/mL of AcLDL for 24 hours demonstrated significant increase in both FC and CE content, 153.72%±21.38% and 240.12%±16.94% respectively compared with control. In the presence of 10 μg/mL of ACATI 58-035, FC content was significantly increased to 198.94%±35.65% over control, while no significant change was observed in CE content (Fig. 4).
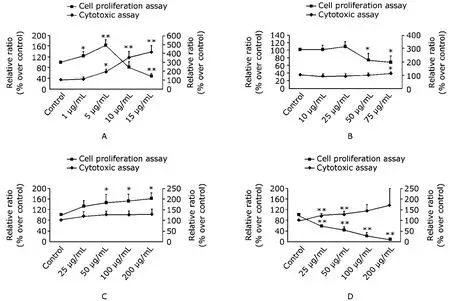
Figure 1. Cytotoxicity of 25OHC, cholesterol, LDL, and AcLDL and their effects on proliferation of human VSMCs. The proliferation results are expressed as ratios of CPM of treated wells over those of controls. The cytotoxicity results are expressed as ratios of readings in OD490nm-690nm of the supernatant over the sum (supernatant plus lysed cells). The left axis represents the relative ratio over control in [3H] incorporation as proliferation index; the right axis is the relative ratio over control in the toxicology assay based on LDH detection. The values are represented as means±SD of quadruplicate wells from four experiments. A. 250HC; B. cholesterol; C. LDL; D. AcLDL. 25OHC: 25-hydroxycholesterol; LDL: low density lipoprotein; AcLDL: acetylated low density lipoprotein; VSMCs: vascular smooth muscle cells; CPM: counts per minute; OD: optical density; LDH: lactate dehydrogenase. *P<0.05, **P<0.001 compared with control.

Figure 2. Morphological changes of human VSMCs after treatment with either or both of 25OHC and ACATI FR 179254. DAPI staining ×400 Human VSMCs were incubated for 24 hours in four different types of media: DMEM containing 0.2% bovine serum albumin (control) (A), 200 nmol/L of ACATI FR 179254 (B), 15 μg/mL of 25OHC (C), and 15 μg/mL of 25OHC plus 200 nmol/L of ACATI FR 179254 (D).

Figure 3.Mo rpho logica l changes of THP-1m acrophages a fter trea tmen twith either or both of AcLDL and ACATIFR 179254.DAPI staining×400 THP-1 cells were differen tia ted by cultu ring in pho rbo l 12-m yristate 13-ace tate(PMA)at a concen tration of 160 nm ol/L for five days in 10%feta l bovine se rum/RPMI 1640 med ium.For the next 24-hou rcu ltu re,the cultu remedium was rep laced by RPMI 1640(con trol)(A),or RPMIw ith 50μg/mL of AcLDL (B),with 100μg/mL of AcLDL (C),or with 50μg/mL of AcLDL plus 800 nm ol/L of ACATIFR 179254(D).
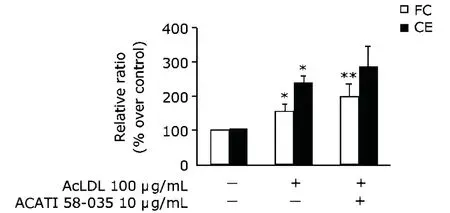
Figure 4.Effect of ACATI on the in trace llu lar free cho leste ro l(FC)and cho leste ro l ester (CE)in AcLDL-loaded THP-1m acrophages.THP-1 cells were diffe ren tiated by cultu ring in PMA at aconcen tration of 160 nm o l/L in 10%FBS/RPMI 1640 m edium for five days, then cultu red for ano ther 24 hou rs in RPMI 1640 med ium(con trol),or in RPMI 1640 with 100μg/mL of AcLDL, or with 100μg/mL of AcLDL plus 10μg/mL of ACATI 58-035.The in trace llu la r FC and CE w ere assayed.The resu lts a re exp ressed as means±SD of the ratios over con tro l(n=3).*P<0.01 com pared with con trol,**P<0.05 com pared with AcLDL treated cells.
D ISCUSSION
Cholesterol and its oxides are believed to be involved in initiation and progression of atherosclerosis.27Many types of cholesterol oxides have been found in atheromatous plaques in hypercholesterolaemic patients.28Kandutsch et al29found that purified cholesterol only had weak inhibitory effect on proliferation of smooth muscle cells (SMCs) in vitro. 25OHC is a much-studied oxysterol that is both formed endogenously and found in the diet. It is also present in atheromatous plaque.3025OHC has been found to in hibit VSMC proliferation in a dose-dependent manne r,and to be a potent inducer of apoptosis i VSMCs which were cultured for 24 hours at the concent rat ion o f 25 μg/m L in 15%FBS.27
Setting 15%FBS/DMEM as con tro l,Yinetal27also demonstrated that cholesterol up to 50 μg/mL did not either inhibit rabbit VSMC proliferation or induce apoptosis,25OHC less than 10 μg/mL did initially stimulate VSMC proliferation, and turned to dose-dependent inhibitory effect thereafter until reaching 50 μg/mL, the maximum concentration tested.
To exclude the interference from the unmeasured lipids in FBS, we used culture medium containing 5% LPDS as control. Our findings coincided with the results of previous studies: cholesterol itself had no inhibitory effect on proliferation of human VSMCs until the concentration exceeded 50 μg/mL. Interestingly, in the concentration range from 1 to 5 μg/mL, 25OHC did stimulate VSMC proliferation,but exerted dose-dependent inhibitory effect at higher concentrations. This will challenge further research on specific concentration-dependent effects of 25OHC on key enzymes in cholesterol turnover at transcriptional and posttranscriptional levels31,32and on consequent distinct cell biology.
The inhibition of proliferation may be explained by several mechanisms. For example, 25OHC may induce the cells to remain in G0, or cause necrosis or apoptosis.Vascular cells possess two major apoptotic pathways commonly referred to as the death receptor pathway and the mitochondrial pathway, both of which could be mobilized in oxysterol-induced apoptosis.33,34What we found based on tha tunde rstanding was that invitro 25OHCinduced apoptosis in human VSMCs was enhanced by ACATI FR 179254.
Foam cells found in vivo have been demonstrated to be of either macrophage or SMC origin.19Acetylation of LDL removes positive charges from the ε-amino groups of lysine and therefore converts a weakly anionic lipoprotein into a strongly anionic one. AcLDL is one of the candidates binding to the scavenger receptors expressed in macrophages, and the non-regulated uptake of modified lipoproteins leads to the formation of foam cells. PMA treated THP-1 cells have been employed as a representative macrophage cell line and used as a model in the study of atherosclerosis and foam cell formation in vitro.35
Apart from the extensive investigation on oxLDL-induced apoptosis in macrophages, some studies have also been conducted on AcLDL-induced apoptosis and produced affirmative results. Nhan et al36found that while about 100% of wild-type peritoneal macrophages harvested from C57BL6/J male mice were TUNEL-positive by 4-day's incubation with 30 μg/mL of oxLDL, AcLDL at the same concentration could also induce a modest increase in TUNEL frequency to approximately 15%. Using double labeling with green fluorescent fluorescein (FITC) and annexin V, Rosenson-Schloss et al37detected the apoptosis in IC21 macrophages cultured in 10% FBS medium containing 25 μg/mL of AcLDL for a maximum of seven hours. In spite of methodological differences, our study also produced evidence of AcLDL-induced apoptosis in PMA fully differentiated THP-1 macrophages by using DAPI staining and caspase assay. Morever, AcLDL-induced apoptosis in THP-1 macrophages was aggravated by ACAT inhibition.
The mechanisms by which ACATI aggravates lipids- induced apoptosis deserve further exploration, suggesting that FC increase might be an important signal in apoptosis initiation. Some previous work established a FC loading model.19,38After intraperitoneal injection of 40 μg concanavalin A in 0.5 mL PBS in adult C57BL/6 female mice for three days, the peritoneal macrophages were harvested and incubated with 100 μg/mL of AcLDL in the presence of 10 μg/mL ACATI 58-035 for five hours. Since cholesterol homeostasis differs between human macrophages and those of murine origin,39,40it was reasonable to investigate whether ACATI 58-035 could also lead to FC loading in AcLDL treated THP-1 macrophages.41The present study provided strong support to the correlation between apo- ptosis in lipid-laden macrophages and FC loading because apoptosis was aggravated by ACAT inhibition and further FC accumulation.
FC accumulation has been detected in apoptotic lipid- laden macrophages, and the functioning target of ACATI was found in ER.42,43Therefore, by directly measuring the sterol content in ER, we found that in AcLDL-induced apoptosis in THP-1 macrophages, there was a significant rise of cholesterol in ER, conforming with the finding of a previous study.44Further research is needed on the mechanism by which ER sterol stress regulating key enzymes and proteins located in ER are involved in cholesterol homeostasis associated with macrophage foam cell formation and apoptosis.
Patients with end-stage renal disease (ESRD) suffer from secondary complex dyslipidemia consisting of both quantitative and qualitative abnormalities in serum lipoproteins resulting from alterations in lipoprotein metabolism and composition.45Atherosclerosis itself is a chronic inflammatory disease; together with disrupted lipid metabolism in chronic kidney disease and inflammatory state, a vicious cycle is formed in ESRD. Therefore, atherosclerosis constitutes the major concern in patients suffering from chronic kidney disease. The present study demonstrated the multiple pathways of atherogenesis, such as LDL-stimulated VSMC proliferation, 25OHC-induced VSMC apoptosis, and AcLDL-induced THP-1 macrophage apoptosis, both of the latter two augmented by ACAT inhibition. The study also confirmed that FC accumulation was a potent inducer of apoptosis, and that sterol stress in ER was the key to initiation of the apoptotic cascade.
1. Moorhead JF, Chan MK, El-Nahas M, et al. Lipid nephrotoxicity in chronic progressive glomerular and tubulo- interstitial disease. Lancet 1982; 2:1309-11.
2. Ruan XZ, Varghese Z, Moorhead JF. An update on the lipid nephrotoxicity hypothesis. Nat Rev Nephrol 2009; 5:713- 21.
3. Ruan XZ, Moorhead JF, Varghese Z. Lipid redistribution in renal dysfunction. Kidney Int 2008; 74:407-09.
4. Littlewood TD, Bennett MR. Apoptotic cell death in atherosclerosis. Curr Opin Lipidol 2003; 14:469-75.
5. Stoneman VE, Bennett MR. Role of Fas/Fas-L in vascular cell apoptosis. J Cardiovasc Pharmacol 2009; 53:100-8. 6. Karaflou M, Lambrinoudaki I, Christodoulakos G. Apoptosis in atherosclerosis: a mini-review. Mini Rev Med Chem 2008; 8:912-8.
7. Isner JM, Kearney M, Bortman S, et al. Apoptosis in human atherosclerosis and restenosis. Circulation 1995; 91:2703-11.
8. Bowen-Pope DF, Schaub FJ. Apoptosis of smooth muscle cells is not silent: Fas/FADD initiates a program of inflammatory gene expression. Trends Cardiovasc Med 2001; 11:42-5.
9. Geng YJ, Libby P. Evidence for apoptosis in advanced human atheroma. Colocalization with interleukin-1 beta- converting enzyme. Am J Pathol 1995; 147:251-66.
10. Kolodgie FD, Narula J, Burke AP, et al. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol 2000; 157:1259-68.
11. Ali YS, Linton MF, Fazio S. Targeting cardiovascular risk in patients with diabetes: management of dyslipidemia. Curr Opin Endocrinol Diabetes Obes 2008; 15:142-6.
12. Fazio S, Major AS, Swift LL, et al. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest 2001; 107:163-71.
13. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 2010; 10:36-46.
14. Tabas I, Weiland DA, Tall AR. Inhibition of acyl coenzyme A:cholesterol acyl transferase in J774 macrophages enhances down-regulation of the low density lipoprotein receptor and 3-hydroxy-3-methylglutaryl-coenzyme A reductase and prevents low density lipoprotein-induced cholesterol accumulation. J Biol Chem 1986; 261:3147- 55.
15. Chang TY, Li BL, Chang CC, Urano Y. Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab 2009; 297:E1-9.
16. Cheng D, Chang CC, Qu X, et al. Activation of acyl-coenzyme A:cholesterol acyltransferase by cholesterol or by oxysterol in a cell-free system. J Biol Chem 1995; 270: 685-95.
17. Warner GJ, Stoudt G, Bamberger M, et al. Cell toxicity induced by inhibition of acyl coenzyme A:cholesterol acyltransferase and accumulation of unesterified cholesterol. J Biol Chem 1995; 270:5772-8.
18. Feng B, Tabas I. ABCA1-mediated cholesterol efflux is defective in free cholesterol-loaded macrophages. Me- chanism involves enhanced ABCA1 degradation in a process requiring full NPC1 activity. J Biol Chem 2002; 277:43271-80.
19. Feng B, Yao PM, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol 2003; 5:781-92.
20. Hatch FT. Practical methods for plasma lipoprotein analysis. Adv Lipid Res 1968; 6:1-68.
21. Fernando RL, Varghese Z, Moorhead JF. Oxidation of low-density lipoproteins by rat mesangial cells and the interaction of oxidized low-density lipoproteins with rat mesangial cells in vitro. Nephrol Dial Transplant 1993; 8:512-8.
22. Goldstein JL, Ho YK, Basu SK, et al. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci U S A 1979; 76: 333-7.
23. Markwell MA, Haas SM, Bieber LL, et al. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal Biochem 1978; 87:206-10.
24. Lowry OH, Rosebrough NJ, Farr AL, et al. Protein measurement with the Folin phenol reagent. J Biol Chem 1951; 193:265-75.
25. Gamble W, Vaughan M, Kruth HS, et al. Procedure for determination of free and total cholesterol in micro- or nanogram amounts suitable for studies with cultured cells. J Lipid Res 1978; 19:1068-70.
26. Heider JG, Boyett RL. The picomole determination of free and total cholesterol in cells in culture. J Lipid Res 1978; 19:514-8.
27. Yin J, Chaufour X, McLachlan C, et al. Apoptosis of vascular smooth muscle cells induced by cholesterol and its oxides in vitro and in vivo. Atherosclerosis 2000; 148: 365-74.
28. Carpenter KL, Taylor SE, Ballantine JA, et al. Lipids and oxidised lipids in human atheroma and normal aorta. Biochim Biophys Acta 1993; 1167:121-30.
29. Kandutsch AA, Chen HW, Heiniger HJ. Biological activity of some oxygenated sterols. Science 1978; 201:498-501. 30. Hodis HN, Crawford DW, Sevanian A. Cholesterol feeding increases plasma and aortic tissue cholesterol oxide levels in parallel: further evidence for the role of cholesterol oxidation in atherosclerosis. Atherosclerosis 1991; 89: 117-26.
31. Edwards PA, Ericsson J. Signaling molecules derived from the cholesterol biosynthetic pathway: mechanisms of action and possible roles in human disease. Curr Opin Lipidol 1998; 9:433-40.
32. Lordan S, Mackrill JJ, O'Brien NM. Oxysterols and mechan- isms of apoptotic signaling: implications in the pathology of degenerative diseases. J Nutr Biochem. 2009; 20:321- 36.
33. Panini SR, Sinensky MS. Mechanisms of oxysterol-induced apoptosis. Curr Opin Lipidol 2001; 12:529-33.
34. Nishio E, Watanabe Y. Oxysterols induced apoptosis in cultured smooth muscle cells through CPP32 protease activation and bcl-2 protein downregulation. Biochem Biophys Res Commun 1996; 226:928-34.
35. Kohro T, Tanaka T, Murakami T, et al. A comparison of differences in the gene expression profiles of phorbol 12-myristate 13-acetate differentiated THP-1 cells and human monocyte-derived macrophage. J Atheroscler Thromb 2004; 11:88-97.
36. Nhan TQ, Liles WC, Chait A, et al. The p17 cleaved form of caspase-3 is present within viable macrophages in vitro and in atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2003; 23:1276-82.
37. Rosenson-Schloss RS, Chnari E, Brieva TA, et al. Glutathione preconditioning attenuates Ac-LDL-induced macrophage apoptosis via protein kinase C-dependent Ac-LDL trafficking. Exp Biol Med (Maywood) 2005; 230: 40-8.
38. Yao PM, Tabas I. Free cholesterol loading of macrophages induces apoptosis involving the fas pathway. J Biol Chem 2000; 275:23807-13.
39. An S, Jang YS, Park JS, et al. Inhibition of acyl-coenzyme A:cholesterol acyltransferase stimulates cholesterol efflux from macrophages and stimulates farnesoid X receptor in hepatocytes. Exp Mol Med 2008; 40:407-17.
40. Waldo SW, Li Y, Buono C, et al. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol 2008; 172:1112-26.
41. Ross AC, Go KJ, Heider JG, et al. Selective inhibition of acyl coenzyme A:cholesterol acyltransferase by compound 58-035. J Biol Chem 1984; 259:815-9.
42. Gill S, Chow R, Brown AJ. Sterol regulators of cholesterol homeostasis and beyond: the oxysterol hypothesis revisited and revised. Prog Lipid Res 2008; 47:391-404.
43. Huang ZH, Gu D, Lange Y, et al. Expression of scavenger receptor BI facilitates sterol movement between the plasma membrane and the endoplasmic reticulum in macrophages. Biochemistry 2003; 42:3949-55.
44. Tao JL, Ruan XZ, Li H, et al. Endoplasmic reticulum stress is involved in acetylated low-density lipoprotein induced apoptosis in THP-1 differentiated macrophages. Chin Med J 2009; 122:1794-9.
45. Vaziri ND. Causes of dysregulation of lipid metabolism in chronic renal failure. Semin Dial 2009; 22:644-51.
 Chinese Medical Sciences Journal2010年2期
Chinese Medical Sciences Journal2010年2期
- Chinese Medical Sciences Journal的其它文章
- Gaussia Luciferase Reporter Assay for Assessment of Gene Delivery Systems in Vivo△
- A Case of Thoracic Spinal Stenosis Secondary to Paget's Disease
- DTNBP1 Gene Is Associated with Some Symptom Factors of Schizophrenia in Chinese Han Nationality△
- Association between the Epidermal Growth Factor Gene and Intelligence in Major Depression Patients△
- Role of Acetylated p53 in Regulating the Expression of map2 in Retinoic Acid-induced P19 Cells△