
牛基因组中新的microRNA预测及分析
2010-01-30 01:32:50钟金城陈智华徐利娟
中国草食动物科学 2010年1期
穆 松,钟金城,陈智华,徐利娟
(西南民族大学动物遗传育种学国家民委—教育部共建重点实验室,成都 610041)
自20世纪90年代以来,作为小分子RNA家族中的一员,MicroRNA(miRNA)已受到广泛的关注,2006年的诺贝尔生理学或医学奖就颁发给了小分子 RNA的研究者。许多研究表明,无论是线虫还是哺乳动物,其体内的miRNA通过分裂或转录抑制目标mRNA达到调控基因表达以及生长发育、细胞增殖和凋亡等作用[1]。
miRNA是长度为20~24个碱基(nt)的非编码单链RNA,广泛存在于真核生物中,其本身不具备开放阅读框(ORF:Open Read Frame)。成熟的miRNA 5'端为一磷酸基团,3'端为羟基,可同上游或下游的序列部分地配对形成茎环结构,miRNA是由具有茎环二级结构的miRNA前体(per-miRNA)加工而成。许多试验表明,miRNA前体在物种间具有高度的进化保守性,其中以茎环部保守性最强。miRNA的表达还具有特异性和时序性特点,在生物体的不同组织中有不同类型的miRNA,生长发育的不同阶段也存在不同种类的miRNA。这些性质都表明miRNA参与了复杂的基因表达调控过程,并决定了生物的生长发育及其行为等变化[2]。
生物信息学技术是预测和发现新miRNA的有效办法[3-5]。大部分的miRNA序列在动物中高度保守,通过计算机软件和其他计算工具可以预测、鉴定出生物的miRNA,并可以利用EST、GSS数据库对miRNA进行大规模的生物信息学预测和分析[6-8]。本研究根据NCBI数据库中的牛EST、GSS信息以及猪、家犬、人、大猩猩和小家鼠5种哺乳动物的已经注册miRNA分子信息,预测牛基因组中新的候选miRNA,以期为进一步寻找牛的miRNA和遗传育种研究提供一条新的思路和方法。
1 材料与方法
1.1 miRNAs、ESTs和 GSSs序列的获得 牛、猪、家犬、小家鼠、大猩猩、人等6种动物的miRNA序列来自于miRBase数据库(http://microrna.sanger.ac.uk/sequences;Release 13.0,March 2009)[9]。牛的EST、GSS[15]和mRNA序列来自于美国国家生物技术信息中心(http://www.ncbi.nlm.nih.gov/)的GeneBank核酸数据库。
1.2 分析软件 序列比对软件为blast-2.2.0-ia32-win32(ftp://ftp.ncbi.nlm.nih.gov/blast/executables/release/2.2.20/blast-2.2.20-ia32-win32.exe),二级结构预测采用RNA structure软件进行,蛋白质序列比对在NCBI提供的Web服务BlastX(http://blast.ncbi.nlm.nih.gov/Blast.cgi)中进行[17]。
1.3 牛基因组中新的miRNAs预测 综合文献[10-11,18]以及通过对牛已经注册的miRNA序列进行分析得到本研究的筛选标准为:①新预测的miRNA与成熟的miRNA只能存在0~3个碱基差异;②新预测miRNA的前体能折叠成发夹二级结构;③发夹结构必须有较小的自由能[14];④miRNA中的A+U含量在30%~70%之间;⑤miRNA与其互补序列的差异不能多于6个;⑥在miRNA中不能存在环状结构。符合以上标准的序列即为本研究所预测到的牛基因组中新的miRNA序列。
2 结果与分析
2.1 牛基因组中新miRNA的预测 根据miRNA高度保守的特点,本研究按照图1所示的思路寻找牛基因组中新的 miRNA。首先下载人、大猩猩、猪、家犬、小家鼠等5个物种的共1 737条miRNA序列,并与牛的已知miRNA进行比对删除相同序列,5个物种间也进行比对删除相同序列,得到了1 445条无重复的目标序列;然后用筛选出的1 445条序列与牛GSS、EST数据库中的序列进行Blast比对,选取其中存在0~3个碱基的同源序列,并删除重复和表达蛋白的序列,最终得到229条序列,其中来自于GSS序列148条、EST序列 81条。
将上述229条序列下载后,选择包含有miRNA相似序列的前后总长共100 nt的片段利用RNAstructure软件进行折叠,观察其二级结构和自由能大小,得到能形成发夹结构且具有较小自由能的共34条序列,其中来自于EST、GSS的序列分别为12条和22条。但是符合A+U碱基含量在30%~70%条件的只有21条序列,来自 EST、GSS的序列数量分别为8条和13条。
将得到的21条miRNA候选序列与sanger数据库per-miRNA分子(http://microrna.sanger.ac.uk/sequences/search.shtml)进行比对,发现同源性较高的有17条,即此17条序列就是本研究预测得到的牛基因组中新的miRNA序列(表1)。
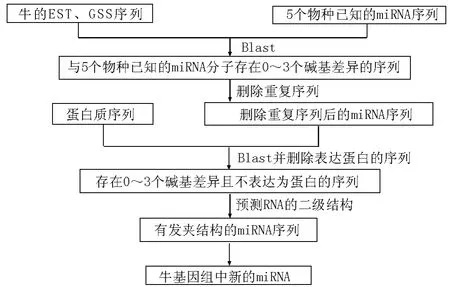
图1 牛基因组中新的miRNA筛选思路
2.2 牛基因组中新miRNA前体的结构特征 由于miRNA序列很短,在基因组中找到匹配序列的概率较大,仅仅搜索相似序列将会产生大量的假阳性结果,如果结合这些相似序列的侧翼序列可能形成二级结构来进一步筛选,能大大减少假阳性率[12]。由图2展示的二级结构可见,本研究得到的17条候选per-miRNAs可以形成发夹结构,包含真正的miRNAs的可能性极大,也表明本研究结果的可靠性。
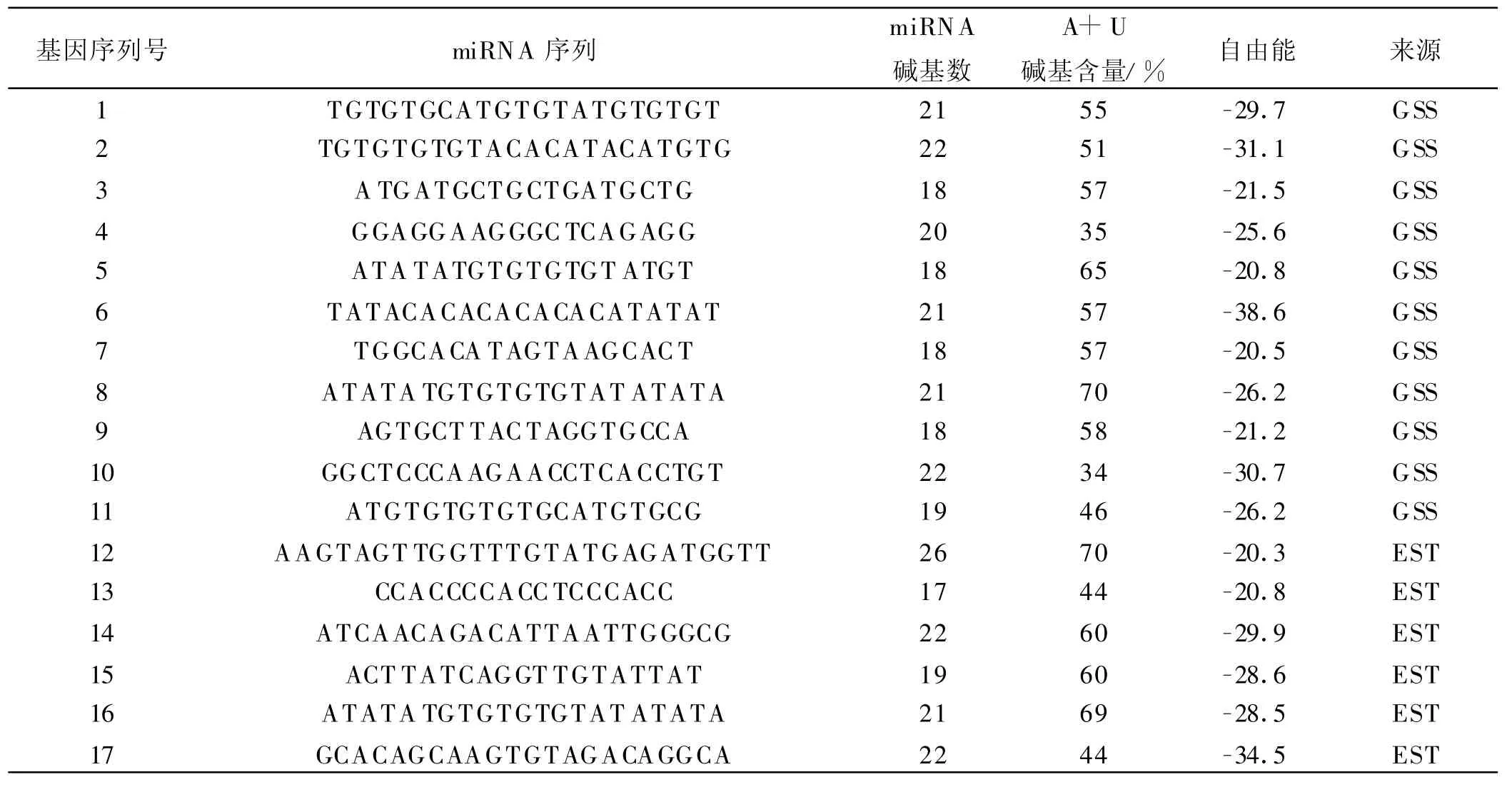
表1 牛基因组中新的miRNA序列
发夹二级结构的形成是miRNA成熟过程中的一个重要步骤,也是miRNA的一个重要特征,但是发夹结构并不是miRNA分子所特有的[13],有些RNAs也能形成类似的发夹结构(mRNA、tRNA、rRNA)。为了避免将其它的RNAs误认为miRNA,本研究引入了自由能(free energy),对已有的per-miRNA统计分析表明,per-RNA具有较小的自由能(表1),说明预测得到的miRNAs符合具有较小自由能的条件。
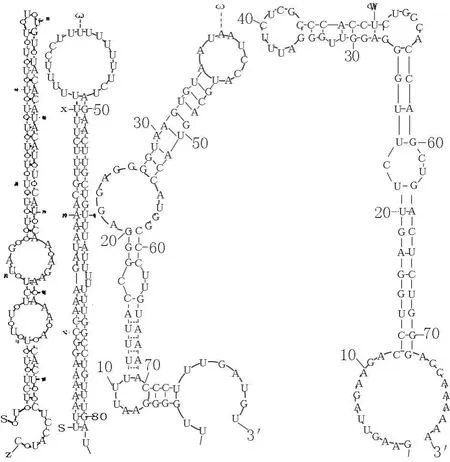
图2 牛基因组中候选miRNA前体分子的二级结构预测
3 讨论
在真核生物中,miRNA具有序列十分保守、前体能折叠成发夹结构、具有较小自由能等特点。本研究得到的17条miRNA,符合miRNA的所有特征,大小在20~24 nt,均能形成发夹状的二级结构,且自由能较小,这表明本研究筛选得到的17条序列可能是牛基因组中新的miRNA,这对牛的遗传育种研究和畜牧业生产具有一定的价值和意义,当然这一结果还需要经过生物学实验的最终验证。
本研究利用生物信息学方法,预测到了17条牛基因组的miRNA候选基因序列,它们都与已知的miRNAs序列高度相似。在生物学实验中,应用芯片技术通过杂交能够发现大量的miRNA分子,但是无法直接得到miRNA前体序列、基因位置和靶基因等信息[16]。但是通过生物信息学方法,除了在对比过程中就能了解前体信息外,还能了解到其靶基因信息,具有许多优越性。这说明根据miRNA的保守性和物种之间基因组的同源性,用生物信息学理论和方法筛选、寻找新的miRNA候选序列的方法能够在较短时间里寻找出一定量的新miRNA分子,速度快、通量大,是一条行之有效的在生物体内寻找到更多miRNA分子的新思路和途径。
经过多年的发展,同源序列搜索的方法已经取得了很大成功,但是本质上需要已知的miRNAs/permiRNAs为参照,搜索与已知miRNAs/per-miRNAs在序列上和结构上同源的 miRNAs/per-miRNAs,对于不与已知miRNAs/per-miRNAs同源的miRNAs/per-miRNAs则无能为力。这也说明生物信息学分析得到的结果是否正确还需要进一步的生物学实验验证,在生物学研究中把生物信息学研究与生物学实验有机地结合是十分重要和必要的。
[1]Bartel D.MicroRNAs,genomics,mechanism and function[J].Cell,2004,116:281-297.
[2]Zarnore P D Haleyb.The big world of small RNAs[J].Science,2005,309:1519-1524.
[3]Zhang B H,Pan X,Cobb G P,et al.Plant micro RNA:A small regulatory molecule with big impact[J].Dve Biol,2006,289:3-6.
[4]Jones-Rhoades M W,Bartel D P.Computational identification of plant microRNAs and their targets,including a stress-induced miRNA[J].Mol Cell,2004,14:787-799.
[5]Guo Z Y,M ao C Q.Computational identification of microRNAs and their targets[J].China Biotechnology,2008,28(10):118-123.
[6]Wang J F,Zhou H,Chen Y Q,et al.Identification of 20 micro RNAs from Oryza sativa[J].Nucleic Acids Res,2004,32:1688-1695.
[7]Arazi T,Talmor-Neiman M,Stav R,et al.Cloning and characterization of microRNAs from moss[J].Plant J,2005,43:837-848.
[8]Zhang B H,Pan X P,Wang Q L,et al.Identification and characterization of new plant microRNAs using EST analysis[J].Cell Res,2005,15:336.
[9]Mathews D H,Sabina J,Zuker M,et al.Ex panded sequence dependences of thermodynamic parameters improves prediction of RNA secondary structure[J].J Mol Biol,1999,288:911-940.
[10]Xie F L,Huang S Q,Guo K,et al.Computational identification of novel microRNA s and targets in Brassica napus[J].FEBS Letters,2008,581:1464-1474.
[11]Qiu C X,Xie F L,Zhu Y Y,et al.Computational identification of microRNAs and targets in Gossypium hirsutum expressed sequence tag s[J].Gene,2007,395(1-2):49-61.
[12]徐德昌,李勇,程大友,等.基于同源搜索的甜菜 MicroRNA计算识别[J].生物信息学,2008,4:152-155.
[13]陈海漩,严忠海,龙建儿,等.应用生物信息学寻找山羊新的 MicroRNA分子及实验验证[J].遗传,2008,30(10):1326-1332.
[14]Zhang B H,Pan X PX,Cox S B,et al.Evidence that miRNAs are different from other RNAs[J].Cell M ol Life Sci,2006,63(2):246-254.
[15]Zhang B,Ban X,Anderson T A.Identification of 188 conserved maize microRNAs and their targets[J].FEBS Letters,2006,580(15):3753-3762.
[16]赵东宇,王岩,罗迪,等.生物信息学中的 MicroRNA预测研究[J].吉林大学学报,2008,5:276-280.
[17]Zuker M.Mflod web server for nucleic acid folding and hybridization prediction[J].Nucleic Acids Res,2003,31:3406-3415.
[18]项安玲,黄思齐,杨志敏.芸苔属植物中 MicroRNA的生物信息学预测与分析[J].中国生物化学与分子生物学报,2008,24(3):244-253.
猜你喜欢
国际公关(2024年2期)2024-03-31 20:31:46
天津医科大学学报(2021年1期)2021-12-05 11:11:05
现代畜牧科技(2021年4期)2021-07-21 06:12:50
天津教育·下(2020年9期)2020-11-16 02:11:22
对外经贸实务(2019年7期)2019-08-06 02:45:53
中国博物馆(2018年2期)2018-12-05 05:28:50
现代检验医学杂志(2016年5期)2016-08-20 03:17:08
湖北师范大学学报(自然科学版)(2015年3期)2015-12-05 03:15:53
现代检验医学杂志(2015年4期)2015-02-06 02:01:55
茶叶通讯(2014年2期)2014-02-27 07:55:40
