
有机硫化物在Ag+改性钛铌酸盐上的吸附和光催化氧化
2010-01-29 02:10何杰,刘娟,李莉
石油学报(石油加工) 2010年2期
何 杰,刘 娟,李 莉
(安徽理工大学化学工程学院,安徽淮南232001)
燃料电池作为最有希望的能源,它使用 H2或碳氢化合物作为燃料。低碳烷烃因 H/C摩尔比高,或直接应用于燃料电池,或通过催化重整、水蒸气变换等过程制备 H2应用于燃料电池。然而这些低碳烷烃中常含有含硫化合物,其对重整催化剂和水汽变换催化剂是毒物,燃料电池的电极材料对硫化物的容许量也非常低。因此,对低碳烷烃作为燃料电池的燃料在使用前必须深度脱硫。
脱除烷烃中硫化物常采用吸附或催化加氢等方法[1]。催化氧化作为深度脱硫方法近年来受到研究者重视[2]。光催化氧化硫化物因具有反应条件温和、工艺简单等特点而成为近年来研究热点之一。光催化氧化不仅可用于有机硫化物的深度氧化,还可对气相中低浓度硫化物进行深度脱除,同时可望在太阳光辐射下进行反应[3-5]。现有的研究表明,使用TiO2或改性 TiO2作为光催化剂,经紫外光辐射一定时间后,有机硫化物最高去除率可达100%。但目前的研究更多地只注意到光催化对有机硫化物氧化的结果,未能探讨烷烃对硫化物的吸附以及光催化过程的影响。笔者以Ag+改性的钛铌酸盐作为催化剂,考察了二甲基硫醚(DMS)和乙硫醇(EM)在其表面上吸附和光催化氧化作用,特别是正己烷对DMS和 EM的吸附和光催化氧化过程的影响,从而进一步探讨烷烃中有机硫化物光催化氧化作用机理。
1 实验部分
1.1 催化剂
将 K2CO3、Nb2O5和 TiO2按6∶5∶1的摩尔比混合,再加入过量(质量分数15%)的 K2CO3,采用高温固相法合成钛铌酸钾盐。2.0 g钛铌酸钾盐用0.5 mol/L的AgNO3溶液25 mL于25℃下处理2 h,洗涤后在相同条件下重复处理1次,洗涤、干燥得Ag+改性的钛铌酸钾盐。
1.2 吸附与光催化
吸附与光催化作用均在静态条件下进行。
1.2.1 DMS、EM吸附和光催化氧化
将0.1 g Ag+离子改性钛铌酸钾盐置于含有DMS(或EM)、分压为13.4 kPa的50 mL密闭容器中,吸附4 h,然后抽真空至1 kPa以去除催化剂表面上弱吸附物种。于室温下在一自制石英反应器(235 mL,φ48 mm×130 mm)中进行光催化反应。0.05 g催化剂置于反应器中,空气气氛,DMS(或 EM)与水汽的分压分别为2.87和3.17 kPa,紫外灯(主波长365 nm,功率300 W)辐射1 h后将体系抽空至1 kPa,所得样品通过红外光谱技术进行表征。
1.2.2 DMS、EM与正己烷混合物吸附和光催化氧化
将0.1 g催化剂置于DMS(或 EM)摩尔分数为3.6×10-5的50 mL正己烷中,室温下搅拌4 h,分离、自然干燥。干燥后的样品分为2部分,一部分直接真空抽脱至1 kPa去除表面弱吸附物种。另一部分置于光催化反应器中,空气气氛中饱和了水蒸气,紫外灯辐射1 h,然后真空抽脱至1 kPa,所得样品通过红外光谱技术进行表征。
1.3 吸附与反应物种表征
采用德国Bruker公司 VECTOR 33傅里叶红外光谱仪对吸附与光催化后的样品进行表征,KBr压片法,扫描范围 4000~400 cm-1,分辨率4 cm-1。
2 结果与讨论
2.1 DMS和 EM在Ag+改性钛铌酸盐表面吸附作用
吸附DMS和EM后的Ag+改性钛铌酸钾盐样品的红外光谱如图1所示。
由图1可见,Ag+改性钛铌酸盐在吸附 DMS前后的 FT-IR谱基本相同,未见二甲基硫的特征峰(1270~1220 cm-1,1440~1415 cm-1)[6],表明它对DMS吸附很弱,经减压解吸即可从Ag+改性钛铌酸盐表面解吸。Ag+改性钛铌酸盐吸附DMS-正己烷混合物后,样品于1232、1319和1159 cm-1出现吸收峰。其中,1232 cm-1吸收峰归属于CH2—S摇摆振动,1319和1159 cm-1分别归属于砜中 SO2不对称伸缩振动和对称伸缩振动[6-7]。可见,非极性溶剂正己烷促进了弱极性DMS在极性Ag+改性钛铌酸盐表面富集。同时,吸附于Ag+改性钛铌酸盐表面的DMS在可见光辐射下被氧化成砜并滞留于其表面。
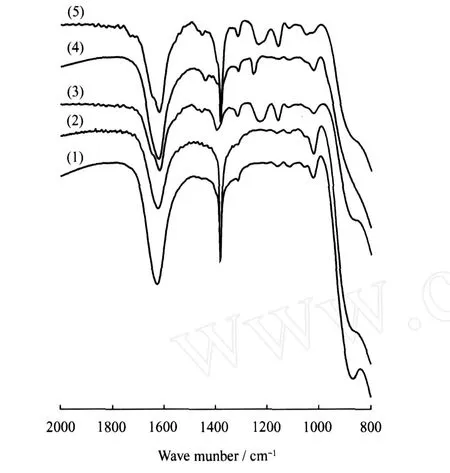
图1 Ag+改性钛铌酸盐吸附DMS和EM前后的FT-IR谱图Fig.1 FT-IR spectra of Ag+-modified titanoniobate before and after adsorbing DMS or EM(1)Fresh Ag+-modified titanoniobate;(2)After adsorbing DMS; (3)After adsorbing mixture of hexane and DMS; (4)After adsorbing EM; (5)After adsorbing mixture of hexane and EM
与DMS不同,Ag+改性钛铌酸盐吸附EM后的FT-IR谱中于1446和1259 cm-1处出现分别归属于CH3弯曲振动和 CH2—S摇摆振动吸收峰,表明其对 EM有较强吸附作用。这种作用可能是EM与 Ag+改性钛铌酸盐中 Ag+作用形成了CH3CH2S-Ag+[8]。吸附EM后的Ag+改性钛铌酸盐样品还于1319 cm-1处出现吸收峰,它归属于SO2的不对称伸缩[6],这是 EM 氧化至乙磺酸C2H5SO2OH的结果。当Ag+改性钛铌酸盐与 EM和正己烷混合物接触后,除 1455 cm-1外,还于1161和1054 cm-1处出现吸收峰,它们分别归属于HOSO2—反对称伸缩振动和对称伸缩振动。可见,正己烷促进了EM在Ag+改性钛铌酸盐表面的吸附和氧化作用。
2.2 吸附在Ag+改性钛铌酸盐表面的DMS和 EM的光催化氧化反应
Ag+改性钛铌酸盐表面分别吸附DMS和 EM并经紫外光辐射后样品的红外光谱如图2所示。
图2(2)显示,吸附于Ag+改性钛铌酸盐表面的DMS经紫外光辐射后,于1205、1123和1034 cm-1出现吸收峰。前者由(CH3O)2SO2产生[6],后二峰归属于单齿硫酸盐[9],表明不仅 DMS分子中的S原子被氧化,而且 C—S键发生断裂,形成了硫酸酯与硫酸盐。图2(3)显示,Ag+改性钛铌酸盐表面吸附DMS-正己烷混合物再经紫外光辐射后,其 FT-IR谱中除了砜(1324和1157 cm-1处SO2反对称伸缩振动和对称伸缩振动)、(CH3O)2SO2和单齿硫酸盐的特征峰外,还于1540和1236 cm-1出现吸收峰,它们归属于表面双齿碳酸盐[9]。与 CH3SCH3、CH3SOCH3和CH3SO2CH3相比,正己烷要稳定得多,且其在 Ag+改性钛铌酸盐表面吸附很弱,因此,表面碳酸盐来自于DMS的深度氧化。说明正己烷促进了DMS在 Ag+改性钛铌酸盐表面吸附,从而导致了DMS的深度氧化。
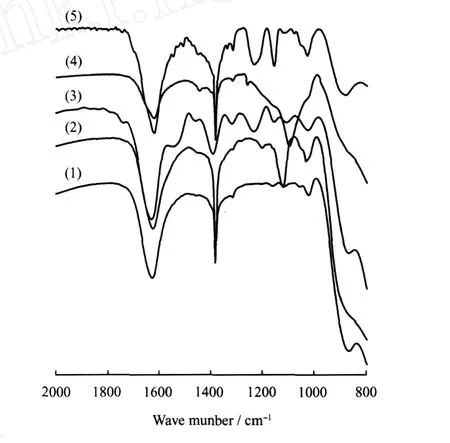
图2 Ag+改性钛铌酸盐表面分别吸附DMS和EM并经紫外光辐射后样品的的FT-IR谱图Fig.2 FT-IR spectra of the samples of Ag+-modifiedtitanoniobate after adsorbing DMS or EM and ultra-light irradiation(1)Fresh Ag+-modified titanoniobate; (2)Ag+-modified titanoniobate adsorbing DMS; (3)Ag+-modified titanoniobate adsorbing mixture of hexane and DMS; (4)Ag+-modified titanoniobate adsorbing EM; (5)Ag+-modified titanoniobate adsorbing mixture of hexane and EM
图2(4)显示,在紫外光辐射下,Ag+改性钛铌酸盐表面吸附的 EM氧化成乙磺酸,于1099 cm-1处出现 HOSO2-对称伸缩振动峰。图中未观察到C—S键断裂形成的氧化产物吸收峰,可能是 EM中C—S键键能高于 DMS中 C—S键键能的缘故[10]。而由图2(5)可见,Ag+改性钛铌酸盐表面吸附DMS-正己烷混合物再经紫外光辐射后,除了—SO2OH振动吸收峰外,还于1120和881 cm-1处出现吸收峰,它们分别归属于单齿硫酸盐和 CO2-3振动吸收,表明 EM发生了深度氧化与矿化,这正是正己烷促进EM在钛铌酸盐表面吸附的结果。
在光催化反应中,由光致空穴同 H2O作用形成的表面 OH自由基具有很强的氧化能力。Vorontsov等[11]认为,DMS氧化主要通过2种途径:C—S键断裂和S原子氧化。S原子氧化生成二甲基亚砜DMSO和二甲基砜DMSO2,C—S键断裂导致DMS氧化成甲醇与磺酸等。这些产物得进一步反应形成了硫酸酯与单齿硫酸盐。王少坤[12]认为,甲硫醇的氧化主要是经过 CH3SH+·OH→CH3S·+H2O反应进行,CH3S·中 S原子被进一步氧化形成磺酸基,这与 EM中C—S键的键能较为一致。存在于混合体系中非极性的正己烷促进了有机硫化物在Ag+改性钛铌酸盐表面上富集,并增强硫化物同催化剂表面相互作用,导致 C—S键减弱,促进了DMS与EM的深度氧化与矿化。
3 结 论
(1)DMS在Ag+改性钛铌酸盐表面吸附较弱,在可见光下稳定。EM与Ag+改性钛铌酸盐表面作用较强,并导致 EM在可见光辐射下氧化。紫外光辐射使DMS和EM均被催化氧化,但因分子中的C—S键稳定性不同,导致氧化途径不同。DMS中C—S键断裂氧化生成硫酸酯与单齿硫酸盐,而 EM的C—S保持稳定。
(2)正己烷促进了DMS和 EM与Ag+改性钛铌酸盐催化剂表面的相互作用,增加了 DMS和EM在催化剂表面的滞留,从而促进了它们在催化剂表面深度氧化。
[1]BABICH I V,MOULIJN J A.Science and technology of novel processes for deep desulfurization of oil refinery streams:A review[J].Fuel,2003,82(6):607-631.
[2]LAMPERT J.Selective catalytic oxidation:A new catalytic approach to the desulfurization of natural gas and liquid petroleum gas for fuel cell reformer applications[J]. Journal of Power Sources,2004,131(1-2):27-34.
[3]赵地顺,刘翠微,马四国.FCC汽油模型化合物光催化氧化脱硫的研究[J].高等学校化学学报,2006,27(4): 692-696.(ZHAO Dishun,LIU Cuiwei,MA Siguo. Oxidation desulfurization from fluid catalytic cracking gasoline vis photocatalysis[J].Chemical Journal of Chinese Universities,2006,27(4):692-696.)
[4]王磊,沈本贤.FCC汽油光化学脱硫反应器模型[J].石油学报(石油加工),2007,21(5):79-84.(WANGLei, SHEN Benxian.The model of FCC gasoline photochemical desulfurization reactor[J]. Acta Petrolei Sinica (Petroleum Processing Section),2007,21(5):79-84.)
[5]NISHIKAWAH,TAKAHARAY.Adsorption and photocatalytic decomposition of odor compounds containing sulfur using TiO2/SiO2bead[J].Journal of Molecular Catalysis A:Chemical,2001,172(1-2):247-251.
[6]The SadtlerHandbook ofInfrared Spectra.Bio-Rad Laboratories Inc,Informatics Division,2004.
[7]GARCIA N G,AYLLON J A,DONENECH X,et al. TiO2deactivation during the gas-phase photocatalytic oxidation of dimethyl sulfide[J].Applied Catalysis B: Environmental,2004,52(1):69-77.
[8]阎武敏,曾勇平,居沈贵.负载Ag+的13X分子筛对乙硫醇和噻吩的吸附性能 [J].石油化工,2006,35(4): 310-313.(YAN Wumin,ZENG Yongping,JU Shengui. Adsorption characteristic of ethanethiol and thiophene on Ag+-13X molecular sieves [J]. Petrochemical Technology,2006,35(4):310-313.)
[9]KOZLOV D V,VORONTSOV A V,SMIRNIOTIS P G,et al.Gas-phase photocatalytic oxidation of diethyl sulfide overTiO2kinetic investigations and catalyst deactivation[J].Applied Catalysis B:Environmental, 2003,42(1):77-87.
[10]JARRIGE J,VERVISCH P.Decomposition of gaseous sulfide compounds in air by pulsed corona discharge[J]. Plasma Chem and Plasma Process,2007,27(3):241-255.
[11]VORONTSOV A V,SAVINOW E V,DAVYDOV L, etal. Photocatalytic destruction of gaseousdiethyl sulfide over TiO2[J].Appl Catal B:Environmental, 2001,32(1):11-24.
[12]王少坤.大气中有机硫化物氧化反应的理论研究[D].山东:山东大学.2002.
猜你喜欢
国际放射医学核医学杂志(2020年2期)2020-05-30
四川冶金(2019年5期)2019-12-23
电线电缆(2016年5期)2016-02-27
中国资源综合利用(2016年7期)2016-02-03
东北电力大学学报(2015年4期)2015-11-13
环境科技(2015年3期)2015-11-08
电源技术(2015年9期)2015-06-05
中国塑料(2014年1期)2014-10-17
应用化工(2014年11期)2014-08-16
应用化工(2014年8期)2014-08-08
