
金属铀环境腐蚀的表面状态研究
2010-01-26 03:47仲敬荣褚明福肖吉群邹乐西
核化学与放射化学 2010年1期
仲敬荣,褚明福,肖 洒,肖吉群,邹乐西
1.中国工程物理研究院,四川 绵阳 621900;2.表面物理与化学国家重点实验室,四川 绵阳 621907
金属铀化学性质活泼,其表面腐蚀行为已引起人们极大关注。已有研究通常采用X(U)PS、AES、S(T)EM、XRD等基于原子水平的表面分析技术,分析铀及铀合金的冶金学及其在不同条件下的腐蚀状态[1-4]。但这些分析方法往往需要(超)高真空环境,且难以获得样品实际贮存环境中腐蚀反应的相关信息。而分子光谱法和扫描探针显微镜技术则能够在关注环境中在线监测材料的反应过程,获取材料表面反应产物的分子结构信息及其微区形貌或电性质变化情况。
分子光谱法主要研究分子中以化学键联结的原子之间的振动光谱和分子的转动光谱,利用特征谱带的频率,推断分子中可能存在的基团或键,进而确定物质的化学结构。红外和拉曼光谱法可看作研究分子能级间跃迁的互补方法,常用于定性和半定量分析。国外已有多个研究者利用激光拉曼、红外光谱等手段实时研究了金属铀[5-6]、铀合金[7]、铀氧化物[8-9]、铀化合物[10]及其表面涂层[11]在不同温湿度和气氛条件下的腐蚀反应,分析推断了各种腐蚀产物及其氧化膜的形成过程,表征出多种离线条件下无法检测到的中间产物。
扫描开尔文力显微镜(scanning Kelvin probe force microscopy,SKPFM)[12-13]作为扫描探针显微镜(scanning probe microscopy,SPM)其中的一种测量模式,与SEM、TEM等其它显微镜技术相比,具有分辨率高(0.1 nm)、实时、实空间、原位成像、环境可控等特点,是材料腐蚀研究的重要手段之一。Kurosaki等[14]利用原子力显微镜(AFM)研究了多晶、单晶(U、Ce)O2的显微结构。Romer等[15]利用电化学原子力显微镜(EC-AFM)在线研究了UO2在不同氧化还原条件和碳酸盐溶液中的微观形貌变化和电化学腐蚀特性。而SKPFM用于铀或其它具有放射性材料的研究则未见公开报道。
本工作拟采用拉曼、红外分子光谱法并结合扫描开尔文力显微镜,在线研究金属铀环境腐蚀的过程和状态,并对不同微区腐蚀产物的组成和形貌进行分析。在此基础上总结铀在大气环境中表面腐蚀的显微形貌及氧化产物的变化规律,分析和评估其表面环境腐蚀的可能机制,有助于深化对铀及其合金环境腐蚀规律的认识,也将为改善防腐措施、提高核燃料的安全可靠性提供有价值的参考信息。
1 实 验
1.1 实验样品
金属铀样品为φ10 mm×2 mm,实验前用砂纸逐级打磨并机械抛光,暂存于无水乙醇中。实验前用体积比1∶1的硝酸溶液超声清洗去除表面氧化皮,使样品表面呈金属银白色光亮镜面,再用去离子水冲洗,无水乙醇清洗2~3次,干燥后立即置于设备反应器中进行分析检测。其它所用试剂均为分析纯。
1.2 实验仪器
Nicolet Almega XR型显微激光拉曼光谱仪(LRS),美国热电公司,532 nm Nd/YAG半导体激光器,100~4 000 cm-1扫描范围,BX51奥林巴斯显微镜,冷热台附件(温度范围:-196~600 ℃,速率:0.1~90 ℃/min);EQUINOX 55型傅立叶变换红外光谱仪(FT-IR),德国布鲁克仪器公司,Globe中红外光源,DTGS检测器,400~4 000 cm-1扫描范围,高温、高压/低真空气-固反应池附件(温度范围:室温~500 ℃,压力不大于2×105Pa,真空低至10-3Pa);SPA 300HV型扫描探针显微镜(SPM),日本精工纳米科技公司,具有接触式AFM、间歇接触式DFM和表面电位SKPFM等多种测量模式。
1.3 实验方法
将高/低温气-固反应池置于拉曼或红外光谱仪的样品室内,连接于自行设计、加工的不锈钢真空系统。仪器光源发出的光经透明视窗入射到反应池内样品表面,热电偶温控仪调节样品反应所需的温度或进行程序升温,检测器收集并记录样品表面反应的光谱信号,经数据处理即可获得试样表面腐蚀产物的特征谱图。结合扫描探针显微镜的SKPFM测量模式,观测样品表面微区在不同实验条件下的形貌变化和电位分布情况。
2 结果和讨论
2.1 金属铀初始形貌及表面状态分析

图1 初始铀样品的金相图Fig.1 Metallographs of original uranium(a,b,c)——明场像(Bright field),(d)——暗场像(Dark field)
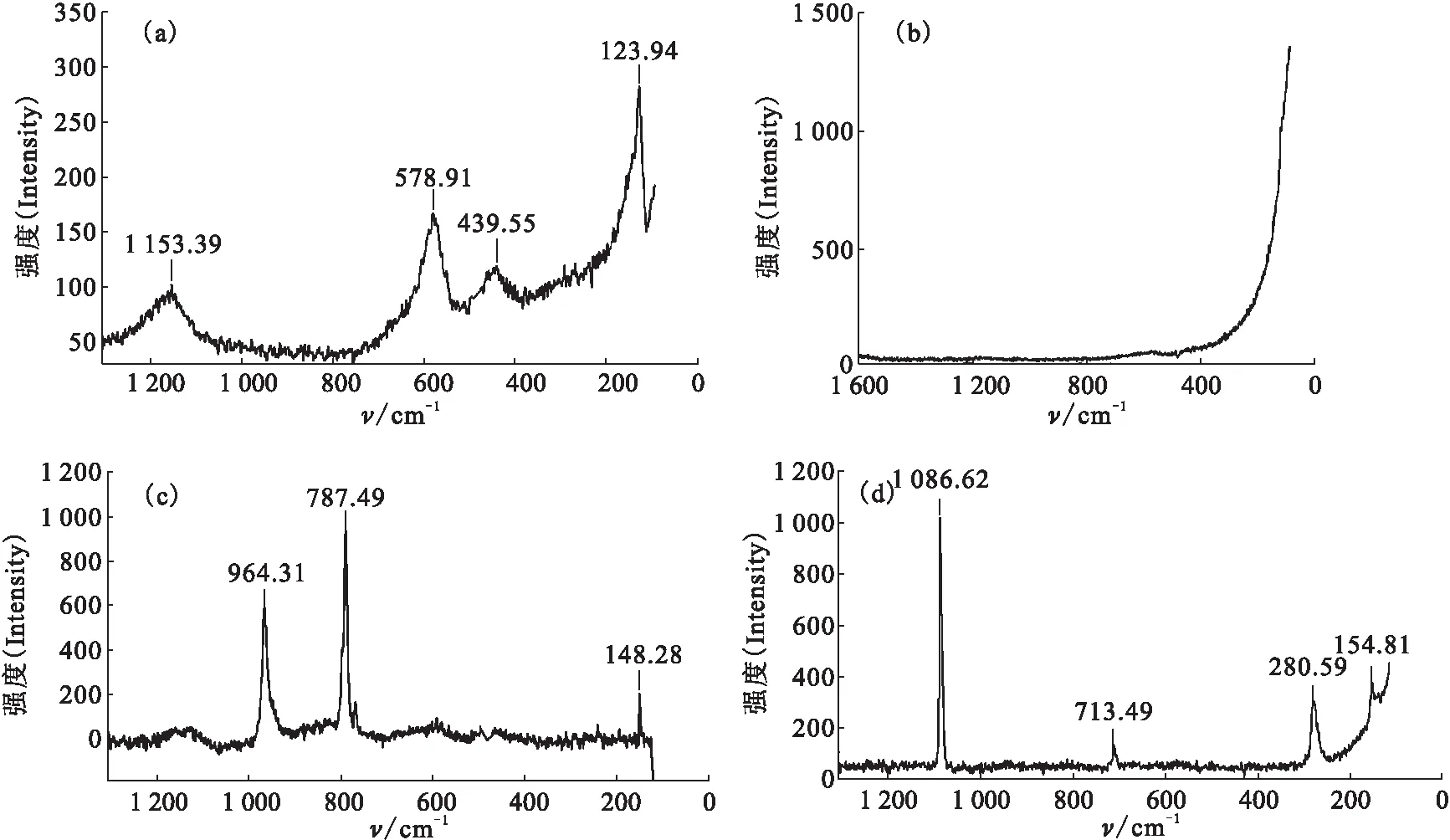
图2 初始铀样品表面不同区域的拉曼光谱图Fig.2 Raman spectra at the different area coverage of original uranium surface(a)——二氧化铀(Uranium dioxide),(b)——碳化铀(UC),(c)——未知铀化物(Unknown compound),(d)——碳酸盐(Carbonates)
然后将金属铀样品置于扫描探针显微镜系统中,利用SKPFM测量模式在大气环境中扫描分析其微区形貌。初始铀样品表面微区(10 μm×10 μm)形貌示于图3。由图3看出,样品表面主要为UO2氧化膜,呈细微粒状不均匀分布,平均粗糙度(Ra)约为8 nm,峰谷高低差(P-V)约为120 nm,其间还夹杂一些大小和高度不等的突起或凹坑,根据拉曼分析结果推断,可能是一些夹杂或表面吸附杂质等。
2.2 金属铀室温腐蚀表面状态分析
(1) 拉曼光谱分析

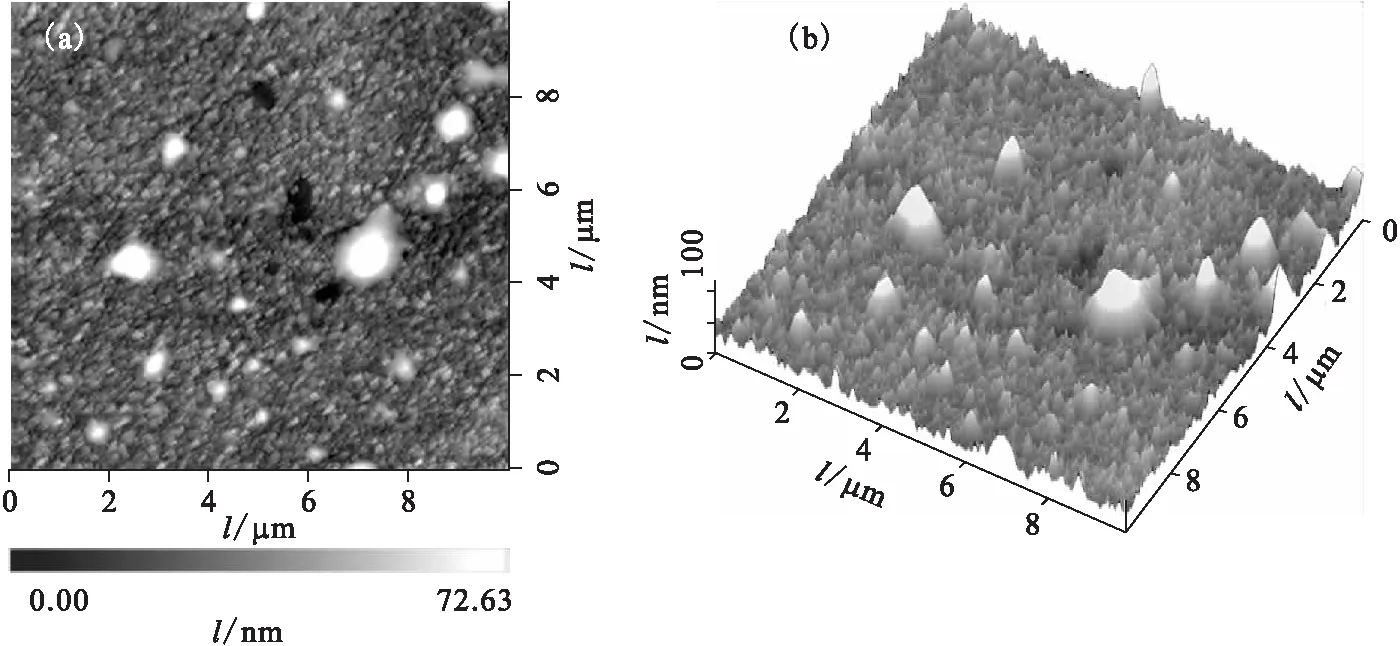
图3 初始铀样品的表面微区形貌图(10 μm×10 μm)Fig.3 Surface microscopic morphologies of original uranium(a)——俯视图(Topographic planform),(b)——三维图(Three-dimensional graph)

图4 空气中放置10 d后铀的表面形貌图Fig.4 Surface microscopic morphology of uranium after 10 days storage in air
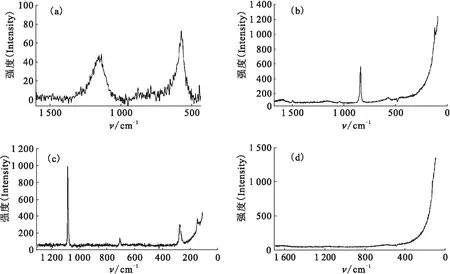
图5 空气中放置10 d后铀表面不同区域的拉曼光谱图Fig.5 Raman spectra at different area coverage of uranium after 10 days storage in air(a)——二氧化铀(Uranium dioxide),(b)——铀酰化物(Uranyl compound),(c)——碳酸盐(Carbonates),(d)——未知UO2+x(Unknown compound)
(2) SKPFM分析
在实验室条件下,将金属铀样品在空气中放置约14 d,样品表面逐渐形成黄色夹杂蓝色和黑色的氧化膜,图6为黑色区域表面微区形貌及对应的电位分布图。由图6看出,铀表面氧化膜(主要为UO2)在形貌图中呈细微球形凸凹粒状不均匀分布,在2 μm×2 μm的扫描范围内,粗糙度约5~8 nm,平均粒径范围100~200 nm,峰谷高低差约50~70 nm,其中还夹杂一些大小不等的凹坑和突起。对应的Kelvin表面电位图显示,样品表面平均电位分布约20~30 mV,电位高低差约350~1 200 mV,且在颗粒边缘和凹坑处的氧化电位较高(400~500 mV),这可用来解释铀及其合金在晶界和缺陷处易发生腐蚀的原因。因为金属铀及其表面铀氧化物常为多晶体,氧化膜内存在大量的晶界和缺陷,而粒子在晶界或缺陷中扩散的活化能远小于晶格内扩散的活化能,与晶格内扩散相比,晶界可看作是氧原子的短程快速扩散通道,易发生吸附氧化,从而造成晶界处氧化电位升高。

图6 金属铀表面黑色氧化膜的形貌图及电位分布图(2 μm×2 μm)Fig.6 Surface microscopic morphology and potential distribution photo of black oxidation film of uranium(a)——形貌俯视图(Topographic planform),(b)——对角线剖面图(Diagonal cutaway view),上为形貌图的对角线剖面图,下为电位图的对角线剖面图(Diagonal cutaway view of topographic planform at the upper side,diagonal cutaway view of surface potential planform at the under side),(c)——表面电位俯视图(Surface potential planform)
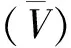
2.3 金属铀热氧化腐蚀的表面状态分析
(1) 拉曼光谱分析
空气中金属铀缓慢升温过程的表面形貌和拉曼谱变化图分别示于图7、8。将金属铀样品在空气中以2 ℃/min的速率缓慢加热至100 ℃,样品表面颜色逐渐变深,同时出现明显的白色腐蚀亮斑(图7(a)),但亮斑点与周围区域的拉曼谱峰并无明显不同,均在576 cm-1和1 156 cm-1有一弱UO2峰。随着温度不断升高,UO2的拉曼谱峰强度明显增加,并逐渐趋于平衡稳定(图7)。进一步升高温度(200~300 ℃),白色腐蚀亮斑逐渐转变为岛状突起,并且区域有所扩大(图7(b))。同时观测到750 cm-1的弱峰,则为U3O8的特征峰,其周围平坦区域仍为UO2的氧化层。随着氧化时间(t)的增加或温度的升高,腐蚀突起不断长大、扩展,并相互连接交错,直至覆盖整个样品表面(图7(c)、图7(d)),各个区域相应的拉曼光谱形状和强度也趋于稳定(图8)。由图8看出,在200 ℃以下的升温过程,铀表面主要氧化生成UO2,升温至230~280 ℃,明显观测到U3O8的特征峰,可以认为UO2向U3O8转变的温度即为260 ℃左右。
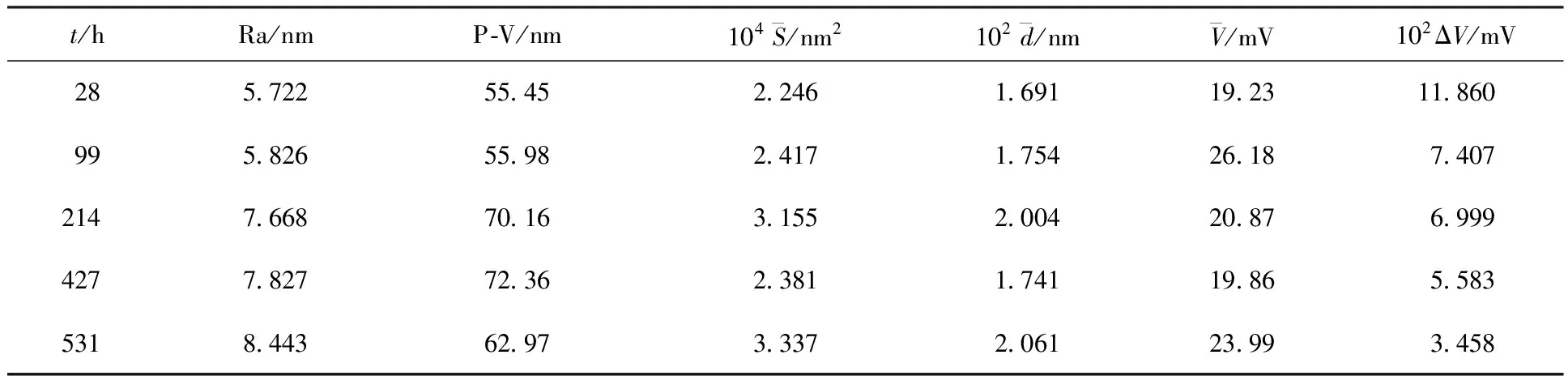
表1 室温条件下金属铀缓慢氧化的表面状态分析结果Table 1 SKPFM analytical results of uranium oxided tardily under ambient conditions
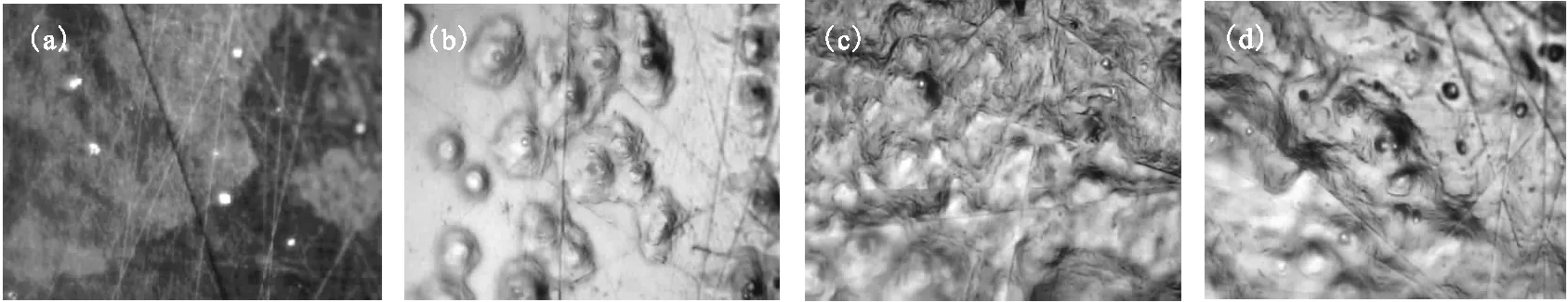
图7 空气中金属铀缓慢升温过程的表面金相图Fig.7 Metallographs of uranium heated slowly in air氧化条件(Oxidation condition):(a)——100 ℃,3 h;(b)——200 ℃,1.5 h;(c)——200 ℃,3 h;(d)——300 ℃,0.5 h
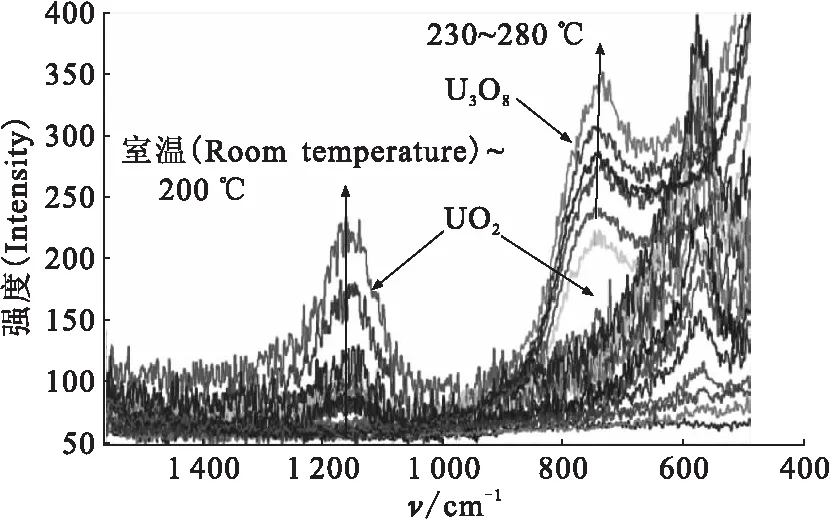
图8 空气中金属铀缓慢升温过程中的拉曼谱变化图Fig.8 Temperature dependence of Raman spectra of uranium heated slowly in air由下至上温度依次为(From the bottom up):室温(Room temperature)、50、100、110、120、130、140、150、160、170、180、190、200、230、240、250、260、270、280 ℃
(2) 红外光谱分析
在实验室环境条件下,将金属铀样品于一定温度范围(50~350 ℃)内连续升温进行热氧化腐蚀实验。不同温度下在线监测样品腐蚀的红外光谱图,获取铀在不同温度下发生化学反应的红外光谱变化信息,结果示于图9。由图9看出,金属铀在100 ℃以上的连续升温过程,迅速发生氧化反应,红外光谱强度逐渐增强。随着温度升高,在570 cm-1附近出现明显的UO2吸收峰,240 ℃以上,谱峰逐渐向高波数范围移动,说明金属铀表面不断氧化生成UO2,还有可能存在非化学计量的UO2+x,260 ℃以上逐渐转变为U3O8,这与拉曼光谱分析结果基本一致。150 ℃金属铀氧化不同时间的红外光谱图示于图10。由图10可知,在150 ℃的恒温过程,随着时间增加,铀表面不断发生氧化腐蚀反应,主要生成UO2(570 cm-1左右)氧化物,约5 h之后,铀表面已形成较厚的氧化层,对红外光形成黑色吸收(完全吸收或散射),表现为UO2的特征红外谱变化不明显。但是,将金属铀由室温迅速升温至200 ℃,铀表面在极短时间内(小于1 h)迅速发生氧化腐蚀,很快形成覆盖整个表面的氧化层,并伴有粉末脱落,主要氧化产物仍为UO2。由此可见,200 ℃以上,铀表面的热氧化腐蚀速率明显高于较低温度时的氧化反应。

图9 空气中不同温度金属铀热氧化腐蚀的红外光谱图Fig.9 FT-IR spectra for thermal oxidation reaction of uranium at different temperature in air由上至下温度如下(From the top down, ordinal temperature):50、100、140、180、200、240、280、300、320、340、350 ℃

图10 150 ℃金属铀氧化不同时间的红外光谱图Fig.10 FT-IR spectra for thermal oxidation reaction of uranium at 150 ℃ in air由上至下氧化条件依次为(From the top down, oxidations):室温(Room temperature),150 ℃:0.0、0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0 h
(3) SKPFM分析
在实验室条件下,将金属铀样品于一定温度范围(室温~200 ℃)内连续升温进行热氧化腐蚀实验。随着时间增长和温度升高,铀表面氧化层不断增厚,不规则的凹坑和突起增多,表面粗糙度增加,颗粒尺寸减小,峰谷高低差增大。同时,铀表面微区的平均电位随温度升高也基本增大,其电位高低差也呈增大趋势。这主要是由于在加热条件下,铀表面氧化膜被破坏,空气中的氧和水气进一步进入基体内部,不断发生腐蚀反应,随着温度升高,表面氧化层开裂并伴有粉末脱落,因此表面电位差增大。
综上所述,金属铀的表面腐蚀反应主要有两个生成阶段。第一阶段是在室温或200 ℃以下富氧条件加热,铀表面主要反应产物是UO2,同时还有可能吸附空气中的O2、H2O和CO2反应生成UO2、铀酰化物、碳酸盐。第二阶段是200~300 ℃的加热过程,铀的表面反应主要是U3O8的成核生长过程[18]。首先U3O8在UO2表面成核,形成一个个“U3O8”岛,并不断向周围长大,这个过程比较缓慢;当样品表面由于U3O8的形成而出现大量破裂时,表面的U3O8沿着晶界和微裂缝快速向内蔓延,呈网状生长,形成一个个表面包裹着U3O8的UO2微粒,这时因氧化比表面积的增加,U3O8出现快速增长,表现为U3O8的拉曼谱峰强度明显增加;当颗粒大部分转化为U3O8后,由于氧化层的增厚,氧原子向颗粒内部扩散变得越来越困难,颗粒内部UO2的进一步氧化变得缓慢,表现为氧化速率转慢,拉曼谱峰的形状和强度也趋于稳定。实验结果与Winer[17]的理论相一致,因此利用表面形貌显微成像和分子光谱识别联用技术,可以更好地在微观上捕捉材料表面不同点位的氧化腐蚀状况,并分析出各点的氧化产物变化信息。
3 结 论
采用显微拉曼、红外以及扫描开尔文力显微镜相结合的分析手段,在线研究了金属铀环境腐蚀的过程和状态,并对不同微区腐蚀产物的组成和形貌进行了分析。其中,金属铀在室温时表面微区形貌呈球形粒状不均匀分布,且颗粒边缘表面电位较高。200 ℃以下富氧条件加热,铀表面的主要氧化产物为UO2,腐蚀从局部反应活性高的点开始;进一步升高温度(200 ℃以上),腐蚀产物由UO2逐渐向U3O8转变,腐蚀突起,不断向周围扩展、长大,最终覆盖整个样品表面,但氧化层疏松,伴有粉末脱落。关于金属铀表面腐蚀的机理及其与光谱强度和表面电位的关系,氧化产物与反应温度、时间及氧分压的定量关系,有待进一步研究。
[1] Sunwoo J L, Anklam T. Uranium Alloy Forming Process Research: UCRL-ID-127172[R]. USA: University of California Lavermore Radiation Laboratory, 1997.
[2] Zabieski C V, Levy M. Fracture Toughness and Stress Corrosion Resistance of U-0.75wt%Ti: ARLTR-200[R]. USA: Army Research Laboratory, 1993.
[3] Manner W L, Lioyd J A, Hanrahan R J. An Examination of the Initial Oxidation of a Uranium-Base Alloy (U-14.1%Nb) by O2and D2O Using Surface-Sensitive Techniques[J]. Appl Surf Sci, 1999, 150: 73-88.
[4] Kelly J, William A L, Manner L. Surface Characterization of Oxidative Corrosion of Uranium-Niobium Alloys: LA-UR-00-4808[R]. USA: Los Alamos National Laboratory, 2000.
[5] Ruan C, Luo W, Wang W, et al. Surface-Enhanced Raman Spectroscopy for Uranium Detection and Analysis in Environmental Samples[J]. Analytica Chimica Acta, 2007, 605: 80-86.
[6] Siekhaus W J. Composition of Uranium Oxide Surface Layers Analyzed by μ-Raman Spectroscopy: UCRL-CONF-201179[R]. USA: University of California, Lavermore Radiation Laboratory, 2003.
[7] Nagelberg A S, Ottesen D K. Corrosion Behavior of Lean Uranium-Titanium Alloys: SAND80-8215[R]. USA: Sandia National Laboratory, 1980.
[8] Yu B Z, Hansen W N. The FTIR Study of Uranium Oxides by the Method of Light Pipe Reflection Spectroscopy[J]. Mikrochim Acta, 1988, I: 189-194.
[9] Caculitan N, Siekhaus W J. The Growth of Epitaxial Uranium Oxide Observed by Micro-Raman Spectroscopy: UCRL-CONF-217799[R]. USA: University of California, Lavermore Radiation Laboratory, 2005.
[10] Lefevre G, Kneppers J, Fedoroff M. Sorption of Uranyl Ions on Titanium Oxide Studied by ATR-IR Spectroscopy[J]. J Colloid Interface Sci, 2008, 327: 15-20.
[11] Roeper D F, Chidambaram D, Halada G P, et al. Development of an Environmentally Friendly Protective Coating for the Depleted Uranium-0.75 wt.% Titanium Alloy Part Ⅳ: Vibrational Spectroscopy of the Coating[J]. Electrochimica Acta, 2006, 51: 4 815-4 820.
[12] Michael R, Florin T. High-Resolution Kelvin Probe Microscopy in Corrosion Science: Scanning Kelvin Probe Force Microscopy(SKPFM) Versus Classical Scanning Kelvin Probe (SKP)[J]. Electrochimica Acta, 2007, 52: 290-299.
[13] Guillaumin V, Schmutz P, Frankel G S. Characterization of Corrosion Interfaces by the Scanning Kelvin Probe Force Microscopy Technique[J]. J Electrochem Soc, 2001, 148(5): B163-B173.
[14] Kurosaki K, Saito Y, Muta H, et al. Nanoindentation Studies of UO2and (U,Ce)O2[J]. J Alloys Compd, 2004, 381: 240-244.
[15] Romer J, Plaschke M, Beuchle G B, et al. In Situ Investigation of U(Ⅳ)-Oxide Surface Dissolutionand Remineralization by Electrochemical AFM[J]. J Nucl Mater, 2003, 322: 80-86.
[16] Schoonover J R, Saab A, Bridgewater J S. Raman/SEM Chemcial Imaging of a Residual Gallium Phase in a Mixed Feed Surrogate[J]. Appl Spectr, 2000, 54: 1 362-1 371.
[17] Winer K A. Initial Stages of Uranium Oxidation: A Surface Study: UCRL-53655[R]. USA: University of California: Lavermore Radiation Laboratory, 1985.
[18] McEachern R J, Choi J W, Kolar M, et al. Determination of the Activation Energy for the Formation of U3O8on UO2[J]. J Nucl Mater, 1997, 249: 58-62.
猜你喜欢
天天爱科学(2022年4期)2022-11-08
辽宁省博物馆馆刊(2021年0期)2021-07-23
中学生数理化·八年级物理人教版(2020年11期)2020-12-14
小学科学(学生版)(2019年11期)2019-12-09
科学大众(2019年8期)2019-10-21
儿童故事画报·发现号趣味百科(2016年7期)2017-02-08
功能材料(2016年10期)2016-11-12
光学精密工程(2016年1期)2016-11-07
当代化工研究(2016年10期)2016-04-11
华北地质(2015年3期)2015-12-07
