
新生儿瓜氨酸血症1 例报道并文献复习
2010-01-24 04:34:46陈玉林余章斌韩树萍邱玉芳董小玥
中国循证儿科杂志 2010年6期
陈玉林 余章斌 韩树萍 朱 春 邱玉芳 沙 莉 董小玥 孙 青
瓜氨酸血症是先天性尿素循环障碍的一种,属常染色体隐性遗传,该病是由于尿素循环中精氨酸琥珀酸合成酶(argininosuccinate synthase,ASS)缺陷所致。国内目前尚未见新生儿瓜氨酸血症存活病例报道,为提高对新生儿瓜氨酸血症的认识,提高早期诊断和治疗水平,本文总结分析1例新生儿瓜氨酸血症的诊断、治疗和随访情况,并对国内外相关病例进行检索和文献复习,分析新生儿瓜氨酸血症病例目前的诊断、治疗、预后和随访现状。
1 病例报道
患儿,男,2 d,因“出汗多6 h”入院,患儿系G3P1,40+6周,生理产娩出,羊水Ⅰ度,Apgar评分10分。生后第2天患儿出现呕吐奶液1次,为非喷射状,予洗胃处理后未再呕吐。入院前16 h患儿出现四肢抽搐1次,头颅B超检查未见明显异常。入院前6 h患儿出现汗多,入院前1 h测血糖7.4 mmol·L-1,伴精神反应欠佳,收入南京医科大学附属南京妇幼保健院NICU治疗。入院查体T 35.5℃,全身皮肤轻度黄染,余未见异常。
1.1 诊治经过 予保暖,补液,混合喂养,查患儿血糖和体温正常,出汗较入院时稍减少但仍偏多。入院后14 h患儿出现喂养时张口困难,四肢肌张力增高,尿量逐渐减少。血气分析示pH 7.50,PCO224 mmHg(1 mmHg=0.133 kPa),PO2113 mmHg,BE -2.9 mmol·L-1,HCO3-18.4 mmol·L-1。电解质分析示Na+143 mmol·L-1,K+4.6 mmol·L-1,Ca2+0.88 mmol·L-1。提示呼吸性碱中毒,低钙血症,给予患儿适当安抚,避免过度通气,补充钙剂。
入院后第2天:凌晨1时许患儿突然出现呼吸暂停伴全身青紫,给予气管插管和皮囊加压给氧后好转。血气分析示pH 7.26,PCO237 mmHg,PO2128 mmHg,BE -10.0 mmol·L-1,HCO3-16.3 mmol·L-1。电解质分析示Na+138 mmol·L-1,K+6.5 mmol·L-1,Ca2+0.85 mmol·L-1。提示代谢性酸中毒,低钙血症,给予纠酸、补液、利尿、多巴胺改善微循环,患儿皮肤逐渐出现花斑纹,呼吸不规则,频繁呼吸暂停,于凌晨4时予机械通气。患儿无自主呼吸;抽搐,予苯巴比妥镇静后缓解,神志不清,反应差;球结膜水肿,瞳孔对光反应迟钝;前囟饱满。查血氨1 692 μmol·L-1(正常参考值9.0~30.0 μmol·L-1),考虑患儿存在氨基酸代谢异常,采血送外院行血串联质谱检查;同时给予静脉滴注盐酸精氨酸,甘露醇脱水处理,禁食。
入院第3天:查血氨2 110 μmol·L-1,肝功能检测:AST 47 U·L-1,ALT 13 U·L-1,肾功能检测:BUN 1.56 μmol·L-1,SCr 34.9 μmol·L-1。限制患儿蛋白摄入量,根据血气分析调整呼吸机参数,对症支持治疗,患儿仍无自主呼吸,反应差。
入院第4天:外院会诊,考虑患儿为先天性遗传代谢性疾病,建议禁食,暂时不予氨基酸、脂肪乳剂。继续予盐酸精氨酸静脉滴注;口服苯甲酸钠和左卡尼汀。继续检测血氨、血气分析,稳定内环境。患儿病情逐渐好转,逐渐下调呼吸机参数,呼吸机治疗4 d后撤离,同时血氨降至75 μmol·L-1,继续给予抗感染治疗。胸部X线和EEG检查均未见异常。
入院第7天:复查肝功能AST 170 U·L-1,ALT 160 U·L-1,总胆红素104.0 μmol·L-1,直接胆红素27.5 μmol·L-1;肾功能指标正常。予复方甘草酸苷护肝治疗。
入院第9天:逐渐开始母乳喂养,奶量逐渐增加,控制蛋白质摄入在1.0 g·kg-1·d-1,口服精氨酸和苯甲酸钠降低血氨。患儿精神发育可,吃奶吸吮可。
入院第12天查血氨降至34 μmol·L-1,应患儿家属要求予以出院。
1.2 辅助检查 患儿出生后第13天回报血串联质谱结果(出生后第4天血样,上海交通大学医学院附属新华医院内分泌、遗传代谢病研究室检测):瓜氨酸438.8 μmol·L-1(正常参考值4~30 μmol·L-1),瓜氨酸/精氨酸134.95(正常参考值 7.0~12.0),提示瓜氨酸血症Ⅰ型。
患儿生后第17天回报血串联质谱结果(出生后第4天血样,北京儿童医院遗传代谢病研究室检测):瓜氨酸2 513.5 μmol·L-1(正常参考值1.79~15 μmol·L-1),瓜氨酸/精氨酸为40.93(正常参考值< 4.33),也提示瓜氨酸血症Ⅰ型(图1)。
患儿生后第29天回报血串联质谱结果(出生后第14天出院时血样,北京儿童医院遗传代谢病研究室检测):瓜氨酸1 040.8 μmol·L-1(正常参考值< 21.3 μmol·L-1),瓜氨酸/精氨酸为66.48(正常参考值< 5.70),提示瓜氨酸水平较前下降(图2)。
患儿生后第33天头颅MRI 检查示(南京儿童医院放射科检测):T1,T2加权成像,可见大脑半球白质信号增高,双侧额颞部脑外间隙增宽;弥散加权成像未见明显信号改变(图3)。
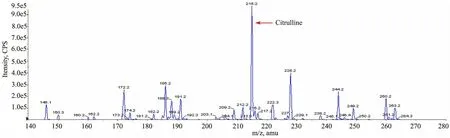
图1 患儿生后第4天血样串联质谱检测结果

图2 患儿生后第14 天血样串联质谱检测结果

图3 患儿生后第33天头颅MRI检查结果
Fig 3 Brain MR image of the patient 33 days after birth
Notes A: T1 weighted image; B: T2 weighted image; C: Diffusion-weighted imaging
1.3 出院后随访 患儿出院后给予正常0~6月龄婴儿奶粉喂养,同时口服盐酸精氨酸 750 mg·d-1和苯甲酸钠750 mg·d-1,患儿一般情况可。出生后第20天盐酸精氨酸和苯甲酸钠均减量至500 mg·d-1,患儿逐渐出现吃奶欠佳,反应差。出生后第29天出现嗜睡和喂养困难,查血氨149 μmol·L-1,予盐酸精氨酸 1.0 g·d-1静脉滴注,连续3 d。出生后第31天查血氨127 μmol·L-1;出生后第33天查血氨18 μmol·L-1;同时查ALT、AST正常;总胆红素和直接胆红素正常;EEG检查表现为爆发性抑制,提示脑功能重度异常。出生后第33天继续予婴儿奶粉喂养,同时口服盐酸精氨酸750 mg·d-1和苯甲酸钠750 mg·d-1。出生后第45天体检,患儿一般情况可,体重4.8 kg,身长58 cm,头围38 cm,均在正常范围。出生后52 d随访,一般情况良好,目前在继续随访中。
2 文献复习
以“新生儿瓜氨酸血症”为检索词检索万方数据库、中国期刊全文数据库和中国维普数据库,以“neonatal citrullinemia” 为检索词检索PubMed数据库,检索文献时间均从建库起至2010年10月。均采用计算机检索,未手工检索灰色文献,中文数据库检索到2篇个案报道[1,2];PubMed数据库共检索到10篇文献[3~12],报道18例患儿。20例瓜氨酸血症特点和预后见表1。

患儿出现症状时间为出生后1~9 d;3例患儿具有遗传代谢性疾病危险因素在出现临床症状前即进行筛查,4例患儿为扩大新生儿遗传代谢性疾病筛查范围时发现,13例患儿出现喂养困难、呕吐、嗜睡、惊厥和昏迷等症状后开始考虑进行遗传代谢性疾病筛查。血瓜氨酸浓度在571~5 950 μmol·L-1;血氨浓度在148~1 941 μmol·L-1。7例在新生儿期死亡,1例在8月龄死亡,6例存活但有神经系统发育障碍,6例存活且神经系统发育、生长发育正常。
3 讨论
瓜氨酸血症根据ASS的异常分为3 型,Ⅰ型(ASS动力学异常)和Ⅲ型(ASS水平低下或无法检测到)也称为经典型(新生儿或婴儿型);Ⅱ型(成人型)几乎均在成年期发病,因肝脏内ASS低下所致[3]。
3.1 临床表现 本病的临床表现主要为呕吐、嗜睡、惊厥和昏迷等。多数患儿出生时无临床表现,24~72 h 后出现呕吐、嗜睡和惊厥等,很快进展为呼吸衰竭和昏迷。国外报道活产婴儿的发病率约为1/57 000,新生儿期病死率极高。国内尚无新生儿瓜氨酸血症发病率的统计。中文数据库中仅检索到2例病例报道,均出现呕吐、嗜睡、惊厥和昏迷等严重临床表现,才考虑采用串联质谱检测确诊。
本文报道的病例因出汗多而入院,入院前患儿只出现四肢抽搐1次,呕吐奶液1次,未出现惊厥和昏迷,此时患儿只是处于新生儿瓜氨酸血症的发病早期。正常情况下,瓜氨酸与天冬氨酸结合形成精氨酸琥珀酸,经过尿素循环转化为尿素排出体外,新生儿瓜氨酸血症患儿ASS合成缺陷,精氨酸琥珀酸不能裂解,瓜氨酸和氨便在体内蓄积,瓜氨酸能增进肝中尿素的形成,有良好的利尿功用,结果早期便会发生出汗多。复习文献报道的20例患儿仅有1例报道多汗的临床表现[7],在没有进行扩大新生儿遗传代谢性疾病筛查范围前,主要是针对有临床表现的高危新生儿进行串联质谱检查,因而新生儿多汗作为新生儿瓜氨酸血症的早期临床表现之一值得注意。
3.2 MRI检查 新生儿尿素循环障碍脑病头颅MRI对评价脑损伤程度及预后有一定的价值。Kara等[4]报道1例新生儿瓜氨酸血症在急性惊厥时,出现脑皮质、皮质下白质受累,基底节正常,脑细胞毒性水肿相关区域出现低弥散系数。如病情继续发展,脑基底节能量耗竭,可受到损害。Majoie等[5]报道1例新生儿瓜氨酸血症,出生后8 d头颅MRI 表现为广泛脑水肿,基底节、丘脑和皮质下白质受损。尹向云等[2]对1例新生儿瓜氨酸血症患儿于出生后14 d行头颅MRI 检查,表现为基底节及皮质下白质均受累。本文报道的病例在出生后33 d行头颅MRI检查,显示大脑半球白质受累,但脑基底节正常,表明该患儿经过治疗延缓了其脑损害的进一步发展,对于其脑损害的远期作用尚需长期随访。
3.3 脑功能检测 由于尿素循环中ASS 的缺陷,导致血、尿及脑脊液中瓜氨酸均升高,尿素循环受阻引起高氨血症[14]。氨的毒性作用是导致神经系统障碍的主要原因,高氨水平导致星形胶质细胞水肿,脑水肿和颅高压[15]。新生儿瓜氨酸血症患儿中枢神经系统的损伤程度取决于血氨水平和高氨持续时间,长时间高氨累积所造成的中枢神经系统损伤是不可逆的。
Clancy 等[10]对3例新生儿瓜氨酸血症恢复期患儿行EEG检查,结果表明患儿血氨浓度和EEG显著异常的(大量的不连续性,爆发抑制,静息电活动)背景活动呈正相关。本文报道的患儿住院期间血氨水平最高为2 110 μmol·L-1,生后第33天EEG检查显示爆发性抑制,提示脑功能重度异常,表明血氨水平与患儿脑损害程度有一定的相关性,而EEG对预后判断的作用值得关注。
3.4 诊断与治疗 新生儿瓜氨酸血症患儿出生时正常,继之出现不明原因多汗、喂养困难、呕吐、抽搐、意识障碍或昏迷等,此时应考虑先天性尿素循环障碍,需及早进行血、尿氨基酸分析进行诊断,并行头颅MRI 或EEG检查以评价脑损伤程度及预后。
该病目前的治疗主要是降低血氨水平,应长期予低蛋白和高碳水化合物饮食,满足机体对每日热量需要即可;可给予精氨酸、苯甲酸钠和苯乙酸钠等降血氨药物;血氨升高显著时可进行血液或腹膜透析。本病为全身性和(或)肝脏中ASS的异常所致,因此大多数的保守治疗效果有限,且要求密切监测代谢产物水平,目前认为肝移植的治疗效果比较确切[15]。
3.5 致病基因 新生儿瓜氨酸血症仅发生于父母双方均携带此缺陷基因的后代,据统计约25%发病,约50%成为基因缺陷携带者。新生儿瓜氨酸血症的致病基因位于第9 号染色体长臂(9q34.1),目前ASS 基因位点已发现有87 种碱基对突变形式[16]。中国这方面的报道较少[17]。
3.6 预后 文献报道的新生儿瓜氨酸血症20例患儿,其中6例存活且神经系统发育、生长发育正常。分析发现,其中3例因为有新生儿瓜氨酸血症高危因素并在无症状时筛查确诊,3例为采用扩大新生儿遗传代谢疾病时筛查并确诊;其余14例患儿出现呕吐、嗜睡、惊厥和昏迷等临床症状后进行串联质谱检测确诊,预后均不良(死亡或有神经系统后遗症)。此外,患儿血氨浓度的高低与神经系统后遗症的严重程度有关。早期筛查、确诊、干预和维持血氨在较低水平是改善患儿预后的主要方法。
3.7 产前诊断 通过检测孕妇妊娠期的代谢物和酶,产前可诊断瓜氨酸血症,一般孕12周经腹绒毛活检术抽吸适量绒毛或孕15~16周取羊水检测瓜氨酸[18]。Kleijer 等[19]25年共检测到36例确诊病例,认为产前诊断瓜氨酸血症是可行的。Chadefaux-Vekemans 等[20]通过检查羊水中瓜氨酸/鸟氨酸+精氨酸的比值来提高检测的准确性(较单纯检测羊水中瓜氨酸的浓度更准确)。此外,通过孕期羊水培养进行ASS 基因位点突变分析也可以确诊胎儿瓜氨酸血症。确诊胎儿于出生后早期给予低蛋白高碳水化合物饮食干预,,可以防止新生儿发生高氨血症造成脑损害,患儿可达到正常的生长发育,已经有成功的产前诊断和新生儿期早期干预的病例报道[21]。
3.8 新生儿筛查 目前开展的新生儿筛查主要针对先天性甲状腺功能低下症(CH)和苯丙酮尿症(PKU)。一些国家已经开始尝试扩大新生儿遗传代谢性疾病筛查的病种,采用串联质谱技术对包括CH和PKU在内的20多种遗传代谢性疾病进行筛查,取得了不错的效果[22]。美国马萨诸塞州从1999年开始进行扩大新生儿遗传代谢性疾病筛查,筛查20万新生儿,发现22例遗传代谢性疾病(发生率为1/10 000),这些新生儿通过早期干预,明显提高了临床预后,未见神经系统后遗症,很少新生儿需要住院治疗[23]。Wilcken 等[24]对通过临床症状诊断和早期扩大新生儿疾病筛查进行比较时发现,从1994至1998年通过临床症状诊断116例遗传代谢性疾病(排除PKU,发生率为7.5/100 000),21例新生儿在出生后5 d死亡或有严重的发育障碍(发生率1.35/100 000);1998至2002年通过早期扩大新生儿疾病筛查诊断70例遗传代谢性疾病(排除PKU,发生率为15.2/100 000),2例新生儿在出生后5 d死亡或有严重的发育障碍(发生率为0.43/100 000)。提示早期扩大新生儿疾病筛查,可提高遗传代谢病患儿的预后。目前,希腊[25]、葡萄牙[25]和美国[27]等国家开展的遗传代谢病筛查均取得了不错的效果。一些研究者进行了早期扩大新生儿疾病筛查项目的筛查、治疗成本-效益分析[28~30],认为扩大新生儿疾病筛查项目值得推广。美国临床生物化学协会对扩大新生儿疾病筛查项目的实施、阳性患儿的治疗和随访提供了规范化的指南[31],值得关注。
本文报道1例新生儿瓜氨酸血症,通过积极治疗,患儿已经存活50 d以上,这是国内报道的第1例存活的新生儿瓜氨酸血症病例,目前患儿生长发育正常,但MRI发现脑白质软化,神经系统发育有待进一步随访评估。遗憾的是,没有得到该患儿ASS 基因突变位点的数据。
[1]Zhang JM (张俊梅),Hou L, Mao J, et al. A report of a case of citrullinemia. J of Chin Physician (中国医师进修杂志), 2009, 11(2):288
[2]Yin XY (尹向云),Xue XD,Fu JH. Congenital urea cycle disorders-a case of neonatal citrullinemia. Chinese Journal of Practical Pediatrics (中国实用儿科杂志), 2010, 25(5):401-402
[3]Kim IS, Ki CS, Kim JW, et al. Characterization of late-onset citrullinemia 1 in a Korean patient: confirmation by argininosuccinate synthetase gene mutation analysis. J Biochem Mol Biol, 2006, 39(4):400-405
[4]Kara B, Albayram S, Tutar O, et al. Diffusion-weighted magnetic resonance imaging findings of a patient with neonatal citrullinemia during acute episode. Eur J Paediatr Neurol, 2009, 13(3):280-282
[5]Majoie CB, Mourmans JM, Akkerman EM, et al. Neonatal citrullinemia: comparison of conventional MR, diffusion-weighted, and diffusion tensor findings.Am J Neuroradiol, 2004, 25(1):32-35
[6]Ohtake A, Takayanagi M, Ogura N, et al. A case of transient neonatal citrullinemia. Eur J Pediatr 1983;141(1):60-61
[7]Nukada O, Uchiyama C, Ubuka S, et al. A case of citrullinemia with abnormal messenger RNA for argininosuccinate synthetase. Acta Paediatr Jpn, 1991, 33(5):672-677
[8]Sanjurjo P, Rodriguez-Soriano J, Vallo A, et al. Neonatal citrullinaemia with satisfactory mental development. Eur J Pediatr, 1991,150(10):730-731
[9]Thoene J, Batshaw M, Spector E, et al. Neonatal citrllinemia: treatment with keto-analogues of essential amino acids. J Pediatr, 1977, 90(2):218-224
[10]Clancy RR, Chung HJ. EEG changes during recovery from acute severe neonatal citrullinemia. Electroencephalogr Clin Neurophysiol, 1991, 78(3):222-227
[11]Melnyk AR, Matalon R, Henry BW, et al. Prospective management of a child with neonatal citrullinemia. J Pediatr,1993, 122(1):96-98
[12]Sander J, Janzen N, Sander S, et al. Neonatal screening for citrullinaemia. Eur J Pediatr, 2003,162(6):417-420
[13]Saheki T, Kobayashi K, Iijima M, et al. Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle. Mol Genet Metab, 2004, 81 (S1):20-26
[14]Sanjurjo P, Rodriguez-Soriano J. Management of neonatal citrullinemia. J Pediatr, 1993, 123(5):838-839
[15]Morioka D, Kasahara M, Takada Y, et al. Current role of liver transplantation for the treatment of urea cycle disorders: a review of the worldwide English literature and 13 cases at Kyoto University. Liver Transpl, 2005, 11(11):1332-1342
[16]Larovere LE, Angaroni CJ, Antonozzi SL, et al. Citrullinemia type I, classical variant. Identification of ASS-p-G390R (c.1168G>A) mutation in families of a limited geographic area of Argentina: a possible population cluster. Clin Biochem, 2009, 42(10-11):1166-1168
[17]Gao HZ, Kobayashi K, Tabata A, et al. Identification of 16 novel mutations in the argininosuccinate synthetase gene and genotype-phenotype correlation in 38 classical citrullinemia patients. Hum Mutat, 2003, 22(1):24-34
[18]Fleisher LD, Harris CJ, Mitchell DA, et al. Citrullinemia: prenatal diagnosis of an affected fetus. Am J Hum Genet, 1983, 35(1):85-90
[19]Kleijer WJ, Garritsen VH, van der Sterre ML, et al. Prenatal diagnosis of citrullinemia and argininosuccinic aciduria: evidence for a transmission ratio distortion in citrullinemia. Prenat Diagn, 2006, 26(3):242-247
[20]Chadefaux-Vekemans B, Rabier D, Chabli A, et al. Improving the prenatal diagnosis of citrullinemia using citrulline/ornithine+arginine ratio in amniotic fluid. Prenat Diagn, 2002, 22(6):456-458
[21]Hong KM, Paik MK, Yoo OJ, et al. The first successful prenatal diagnosis on a Korean family with citrullinemia. Mol Cells, 2000,10(6):692-694
[22]Waisbren SE, Albers S, Amato S, et al. Effect of expanded newborn screening for biochemical genetic disorders on child outcomes and parental stress. JAMA, 2003, 290(19):2564-2572
[23]Marsden D. Expanded newborn screening by tandem mass spectrometry: the Massachusetts and New England experience. Southeast Asian J Trop Med Public Health, 2003, 34(S3):111-114
[24]Wilcken B, Haas M, Joy P, et al. Expanded newborn screening: outcome in screened and unscreened patients at age 6 years. Pediatrics, 2009, 124(2):241-248
[25]Loukas YL, Soumelas GS, Dotsikas Y, et al. Expanded newborn screening in Greece: 30 months of experience. J Inherit Metab Dis, 2010, in press
[26]Vilarinho L, Rocha H, Sousa C, et al. Four years of expanded newborn screening in Portugal with tandem mass spectrometry. J Inherit Metab Dis, 2010, in press
[27]Centers for Disease Control and Prevention (CDC). Impact of expanded newborn screening-United States, 2006. MMWR Morb Mortal Wkly Rep, 2008, 57(37):1012-1015
[28]Wilcken B. Expanded newborn screening: reducing harm, assessing benefit. J Inherit Metab Dis, 2008,33(S2):205-210
[29]Prosser LA, Kong CY, Rusinak D, et al. Projected costs, risks, and benefits of expanded newborn screening for MCADD. Pediatrics,2010,125(2):286-294
[30]Cipriano LE, Rupar CA, Zaric GS. The cost-effectiveness of expanding newborn screening for up to 21 inherited metabolic disorders using tandem mass spectrometry: results from a deci-sion-analytic model. Value Health, 2007, 10(2):83-97
[31]Dietzen DJ, Rinaldo P, Whitley RJ, et al. National academy of clinical biochemistry laboratory medicine practice guidelines: follow-up testing for metabolic disease identified by expanded newborn screening using tandem mass spectrometry; executive summary. Clin Chem, 2009, 55(9):1615-1626
猜你喜欢
中老年保健(2022年2期)2022-11-25 23:46:31
生命科学研究(2022年2期)2022-05-15 08:02:56
昆明医科大学学报(2022年1期)2022-02-28 07:46:54
首都食品与医药(2021年5期)2021-03-22 11:25:04
转化医学电子杂志(2018年11期)2018-01-16 12:37:42
老年医学与保健(2017年6期)2017-02-06 05:29:45
现代检验医学杂志(2016年1期)2016-11-12 13:19:34
中国现代药物应用(2016年10期)2016-03-06 07:18:45
中国免疫学杂志(2016年2期)2016-01-30 21:18:21
疑难病杂志(2014年12期)2014-04-16 05:19:33
