
我国茶毛虫及其布尼亚病毒(EpBYV)的遗传多样性分析
2024-11-08 00:00:00陈世春江宏燕廖姝然陈亭旭牛金志王晓庆
茶叶科学 2024年5期
摘要:茶毛虫是一种危害茶叶生产的重要害虫和人类皮肤致敏原,布尼亚病毒在茶毛虫的不同地理种群中广泛分布,研究茶毛虫与茶毛虫布尼亚病毒(Euproctis pseudoconspersa bunyavirus,EpBYV)遗传背景可为更好地防治茶毛虫和开发利用病毒资源提供理论支撑。采集了15个地理种群共148个茶毛虫幼虫样本,测定了茶毛虫的COI、ND5序列及EpBYV的RdRp序列,利用软件DnaSP 6.12.03、Arlequin 3.5.2.2和MEGA 7.0.26等进行遗传多样性分析。合并COI、ND5序列分析发现,茶毛虫的15个地理种群具有较高的单倍型多样性(h=0.880 68)和较低的核苷酸多样性(π=0.003 17),99个种群对间遗传分化显著较高(FST>0.290,P<0.05),AMOVA分析显示遗传变异主要来自于种群之间(87.12%),且组间分化与我国地势第二、三级阶梯分界吻合,种群历史动态分析推测茶毛虫总群体在近期较为稳定。EpBYV的RdRp序列在除重庆城口种群外的138个样本中成功扩增,具有较高的单倍型多样性(h=0.935 26)和相对较低的核苷酸多样性(π=0.017 95),93个种群对间遗传分化显著较高(FST>0.257,P<0.05),遗传变异主要来自于种群之间(62.13%),种群历史动态分析支持EpBYV种群近期经历了种群扩张事件。综合研究结果表明,我国茶毛虫总群体在近期将较为稳定,但在重庆城口和福建宁德有种群扩张风险;EpBYV群体经历了种群扩张,EpBYV在茶毛虫群体中的感染率和种群扩张能力都较高,具有应用于茶毛虫生物防治的潜力。
关键词:茶树;茶毛虫;布尼亚病毒;EpBYV;遗传多样性
中图分类号:S571.1;S435.711 文献标识码:A 文章编号:1000-369X(2024)05-793-14
Genetic Diversity Analysis of Euproctis pseudoconspersa and Its Bunyavirus (EpBYV) in China
CHEN Shichun1, JIANG Hongyan1, LIAO Shuran1, CHEN Tingxu1, NIU Jinzhi2, WANG Xiaoqing1*
1. Tea Research Institute of Chongqing Academy of Agricultural Sciences, Chongqing 402160, China;
2. College of Plant Protection, Southwest University, Chongqing 400715, China
Abstract: Tea tussock moth, Euproctis pseudoconspersa, is an important pest which damages tea plants and causes human dermatitis. Euproctis pseudoconspersa bunyavirus, EpBYV, is a bunyavirus that widely distributed in different geographical populations of E. pseudoconspersa. In order to control the E. pseudoconspersa and utilize the virus resources, it is necessary to fully understand the genetic background of E. pseudoconspersa and EpBYV. In this study, 148 samples of E. pseudoconspersa larvae from 15 geographic populations were collected. COI and ND5 gene sequences of E. pseudoconspersa and RdRp sequences of EpBYV were determined. The genetic diversities of E. pseudoconspersa and EpBYV were analyzed by DnaSP 6.12.03, Arlequin 3.5.2.2 and MEGA 7.0.26. Sequence analysis of the combined sequences of COI and ND5 genes shows that 15 geographic populations have high haplotype diversity (h=0.880 68) and low nucleotide diversity (π=0.003 17). Significantly high genetic differentiation among 99 population pairs (FST>0.290, P<0.05) was identified. Molecular variance analysis (AMOVA) shows that the genetic differentiation of E. pseudoconspersa was mainly among populations (87.12%), and the differentiation among groups was consistent with the second and third ladder boundaries in China. Demographic history analysis suggests that the population of E. pseudoconspersa is relatively stable. RdRp sequences were successfully amplified in the 138 samples except CK population. RdRp sequence analysis reveals that the 14 geographic populations of EpBYV had high haplotype diversity (h=0.935 26) and relatively low nucleotide diversity (π=0.017 95). The 93 population pairs had significantly higher genetic differentiation (FST>0.257, P<0.05). AMOVA analysis shows that the genetic differentiation of EpBYV was mainly between populations (62.13%). Demographic history analysis reveals that EpBYV might have undergone population expansions in the past. Based on the analysis of this study, the population of E. pseudoconspersa in China is relatively stable, and there is a risk of population expansion in Chengkou, Chongqing and Ningde, Fujian. The population of EpBYV has experienced population expansion. The infection rate and population expansion ability of EpBYV in E. pseudoconspersa are high, which has good potential for biological control of E. pseudoconspersa.
Keywords: tea plant, Euproctis pseudoconspersa, bunyavirus, EpBYV, genetic diversity
茶毛虫(Euproctis pseudoconspersa Strand),又名茶黄毒蛾,隶属于鳞翅目(Lepidoptera)毒蛾科(Lymantriidae),是一种遍布我国各大茶区的重要害虫,以幼虫咀食叶片为害,严重时可致茶丛光秃,造成茶叶产量严重受损,且各虫态的茶毛虫毒毛触及人体均易引起皮炎,严重影响茶园农事活动及周边居民健康[1-5]。昆虫病毒广泛应用于鳞翅目害虫的防治[6-7],目前茶毛虫的病毒研究和应用主要是茶毛虫核型多角体病毒(Euproctis pseudoconspersa nuclear polyhedrosis virus,EpNPV)[8-11],其苏云金杆菌(Bacillus thuringiensis,BT)混剂已实现了商品化[12-14]。EpNPV对茶毛虫控制效果较好,但在田间调控存在滞后、株系间毒力差异[8],以及不同地理种群茶毛虫对同株EpNPV敏感性不同等问题[15]。进一步收集并研究其他病毒,对茶毛虫生物防治资源的储备及生物防治效果的增强具有重要意义。茶毛虫布尼亚病毒(Euproctis pseudoconspersa bunyavirus,EpBYV)属于布尼亚病毒目(Bunyavirales)白纤病毒科(Phenuiviridae),是一种新鉴定的感染茶毛虫的负链RNA病毒[16]。已有研究证实,布尼亚病毒目的费拉病毒(Ferak virus,FERV)可通过蚊子产卵进行垂直传播,并可能具有影响宿主传播其他病毒的能力[17-19]。茶毛虫感染EpBYV虽无明显感染症状,但EpBYV可在茶毛虫体内复制并诱发体内的RNAi抗病毒免疫反应[15]。因此,研究EpBYV可为构建基于RNA病毒的茶毛虫绿色防控体系提供基础数据。
茶毛虫作为一种全国茶区均有分布且在部分茶区屡有暴发的害虫,近年来对其研究主要集中在田间防控方面[20-21]。对茶毛虫开展遗传多样性研究,可更全面地从分子水平了解其种群遗传结构,解析其生态适应机制和成灾规律,为各茶区制定有效的防控策略提供基础信息。此前,已有学者对茶小绿叶蝉[22]、茶网蝽[23]、茶棍蓟马[24]开展了相关研究。因此,本研究采集了我国四大茶区11个省份共15个地区茶园的茶毛虫幼虫样本,扩增茶毛虫的线粒体COI和ND5序列,以及EpBYV的RNA聚合酶(RNA-dependent RNA polymerase,RdRp)编码基因序列,联合分析茶毛虫和EpBYV的种群遗传多样性和遗传结构特征,探讨茶毛虫种群历史动态,评估其近期在全国茶区的成灾风险,明确EpBYV对茶毛虫的感染率、遗传变异和种群发展动态历史,评估其未来用于生物防治的潜力。
1 材料与方法
1.1 试验材料
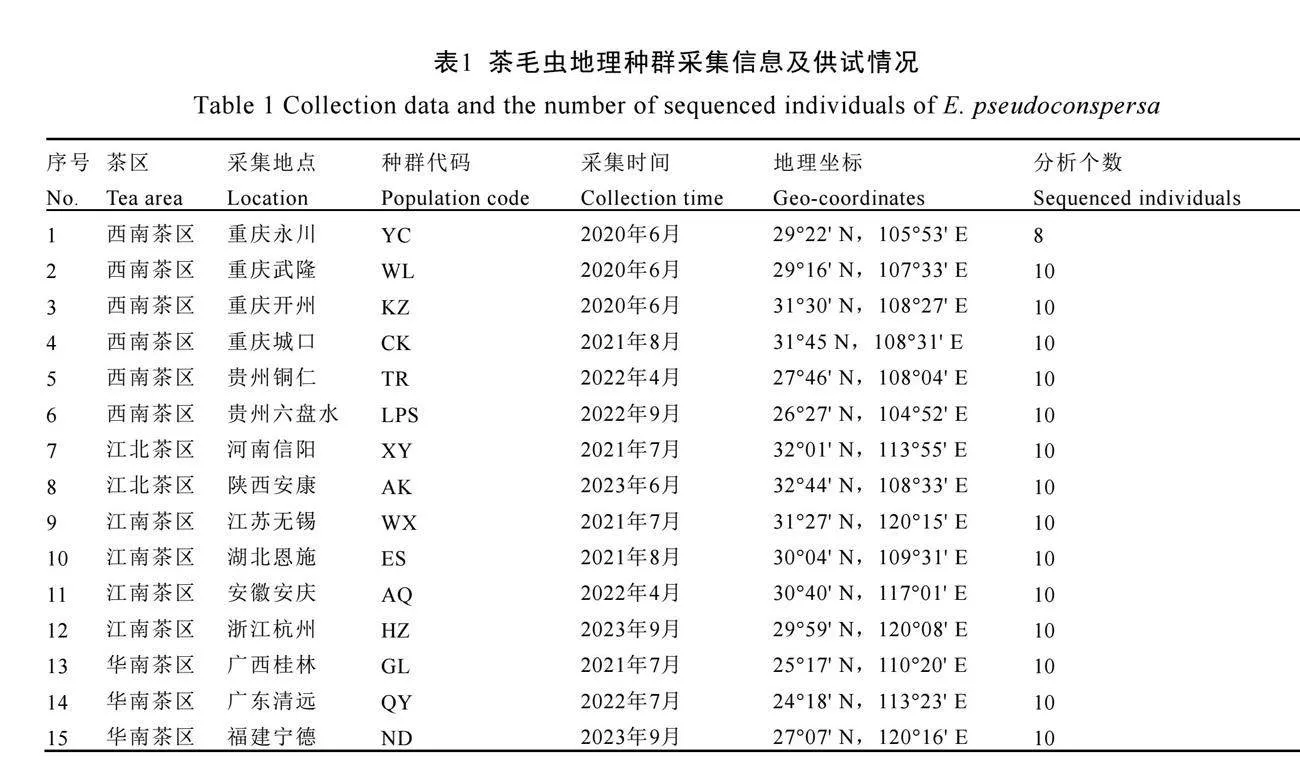
试验所用茶毛虫幼虫样本于2020年6月至2023年9月采集自我国四大茶区(江南茶区、江北茶区、西南茶区、华南茶区)11个省份的15个茶园,具体信息如表1所示。
1.2 试验方法
1.2.1 DNA、RNA提取及cDNA合成
取PBS缓冲液清洗后的茶毛虫幼虫,纵切后取半头经液氮速冻、研磨,使用血液/细胞/组织基因组DNA提取试剂盒[天根生化科技(北京)有限公司]提取DNA,﹣20 ℃保存,用于茶毛虫线粒体基因序列扩增;取另半头茶毛虫经速冻研磨后采用TRIzol法提取总RNA,使用RQ1 RNase-Free DNase试剂盒(Promega)去除RNA样品中的基因组DNA,再使用PrimeScriptTM 1st Strand cDNA Synthesis Kit试剂盒(TAKARA)反转录合成cDNA,﹣20 ℃保存,用于EpBYV的RdRp序列扩增。
1.2.2 PCR扩增及测序
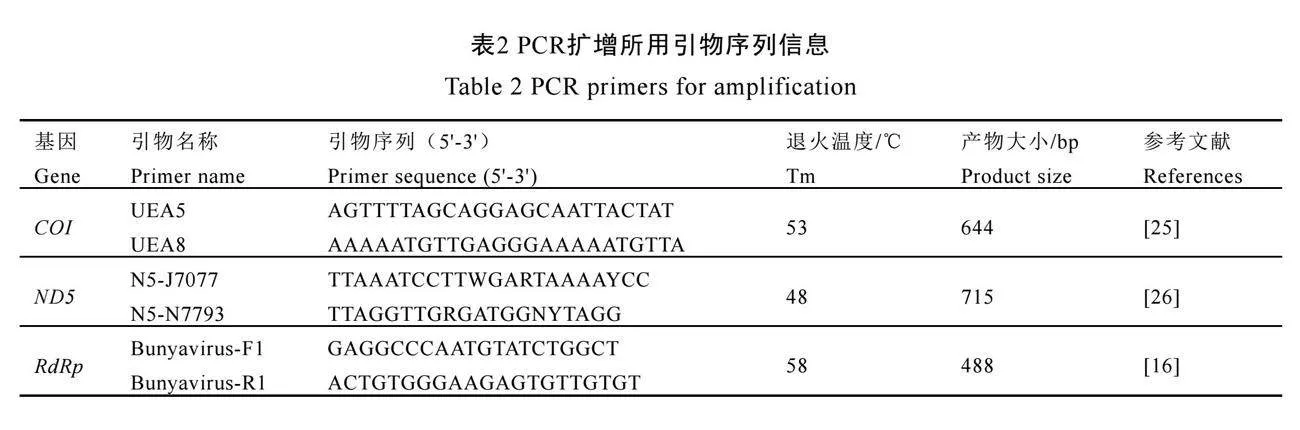
茶毛虫线粒体基因COI、ND5,以及EpBYV病毒中RdRp序列的PCR扩增引物如表2所示。使用Taq HS DNA聚合酶(TaKaRa)进行PCR扩增,总反应体积为25 μL。扩增条件为95 ℃预变性3 min;95 ℃变性30 s,按表2中的退火温度退火30 s,72 ℃延伸1 min,34
个循环;72 ℃终延伸10 min。扩增完成后取2 μL PCR产物进行1%的琼脂糖凝胶电泳,检测成功后将PCR产物送至北京六合华大基因有限责任公司进行双向测序。测序得到的序列使用DNAMAN软件拼接,序列翻译后在NCBI数据库中进行BLAST比对,验证序列的准确性。
1.2.3 数据整理与分析
利用DnaSP 6.12.03软件[27-28]计算茶毛虫不同地理种群的单倍型多样性(Haplotype diversity,h)、核苷酸多样性(Nucleotide diversity,π)等遗传多样性指标。基因流(Nm)依据公式Nm=(1-FST)/4FST计算[29]。在Arlequin 3.5.2.2软件[30]中使用Tajima’s D[31]和Fu’s Fs[32]统计进行中性检验,并进行种群的分子遗传变异方差分析(Analysis of molecular variance,AMOVA),计算种群对间的遗传分化指数FST和误配等。基于Kimura 2-Parameter模型使用MEGA 7.0.26软件[33]计算种群间的遗传距离。采用Network 10.2软件[34]绘制单倍型之间的网络进化图(Median-joining network),单倍型的最大似然法系统发育树使用PhyML3.0(www.atgc-montpellier.fr/phyml)构建[35]。
2 结果与分析
2.1 序列变异和遗传多样性
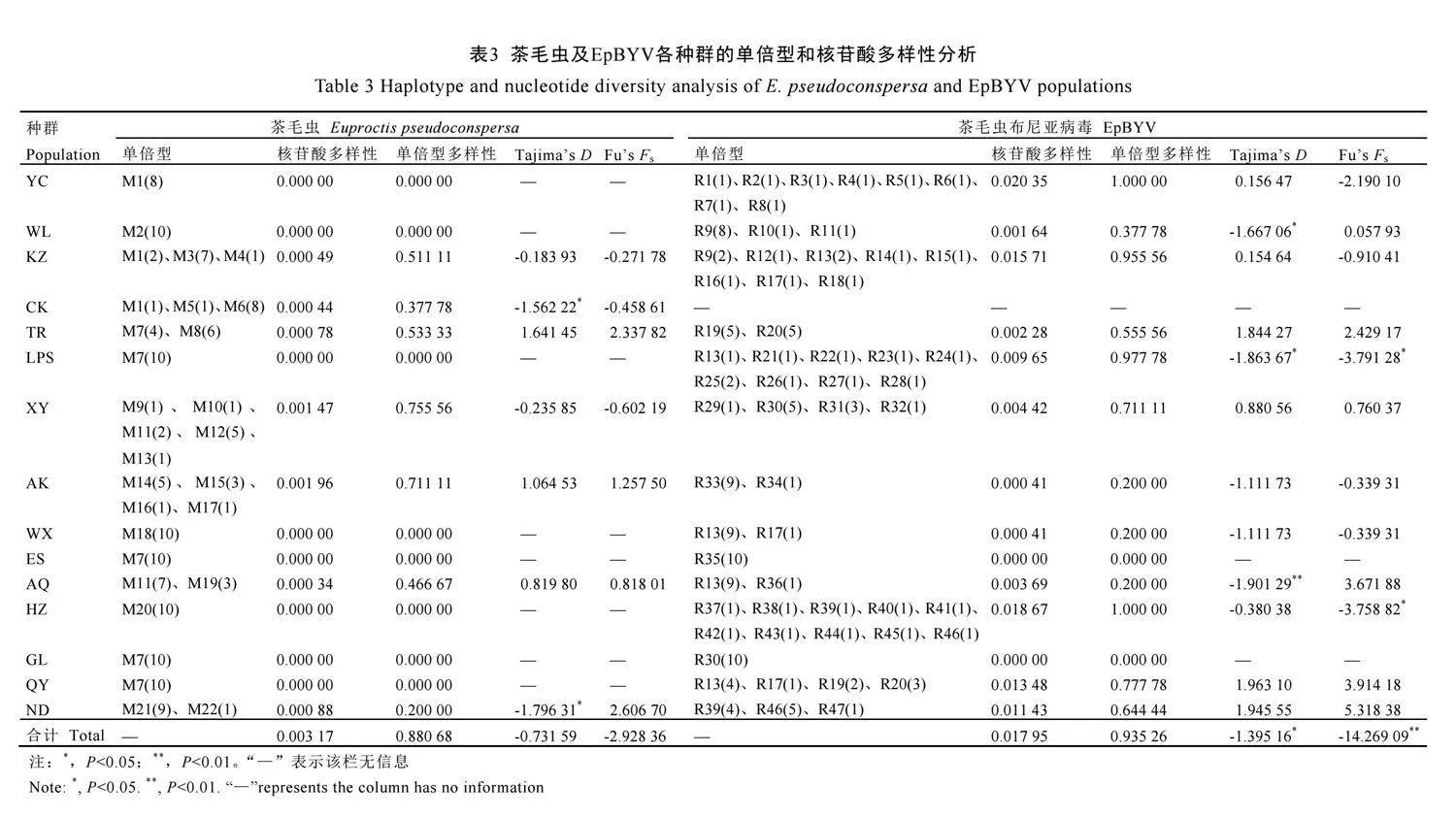
本研究共采集到15个地理种群共148头茶毛虫幼虫,均扩增获得644 bp的COI序列和715 bp的ND5序列。将同一样本的两个序列片段分别合并,获得148条长度为1 359 bp的序列,其中保守位点1 327个,变异位点32个,未发现碱基缺失或插入现象。在32个变异位点中自裔位点5个,简约信息位点27个,均为两碱基变异位点,共含有22个单倍型(表3),将M1中的COI和ND5序列上传GenBank,获得登录号分别为PQ097955和PQ096005。遗传多样性分析显示(表3),总群体的核苷酸多样性指数为0.003 17,单倍型多样性指数为0.880 68。15个地理种群中,永川、武隆、六盘水、无锡、恩施、杭州、桂林、清远等地的8个种群不具有多态性位点,各项指数均为0,表明其种群内部序列保守,遗传多样性极低;其余7个种群的核苷酸多样性指数为0.000 34~0.001 96,单倍型多样性指数为0.200 00~0.755 56,各种群间遗传多样性差异较为明显,其中信阳和安康两个种群的遗传多样性较高。
EpBYV的RdRp序列扩增结果显示,除重庆城口10个样本外,其他138个样本均成功获得488 bp的RdRp序列,扩增成功率为93.24%。获得的138条RdRp序列中均无碱基缺失或插入现象,其中保守位点有402个,变异位点86个,约占全长的17.62%,无碱基缺失或插入现象。86个变异位点中有自裔位点49个,简约信息位点37个,其中两碱基变异位点82个,三碱基变异位点4个,形成单倍型47个(表3),将R1序列上传GenBank获得登录号为PQ096006。遗传多样性参数显示(表3),总群体的核苷酸多样性指数(0.017 95)和单倍型多样性指数(0.935 26)均大于前文中茶毛虫遗传多样性的相应指标。相对于茶毛虫线粒体序列无突变位点的8个种群,EpBYV的RdRp序列仅在桂林和恩施种群中无突变位点,表明EpBYV种群内部的遗传多样性较高。13个EpBYV有突变位点的种群中,核苷酸多样性指数为0.000 41~0.018 67,单倍型多样性指数为0.200 00~1.000 00,其中杭州和永川两个种群的单倍型多样性指数均为1.000 00,遗传多样性最高,对应的每个样本单独成为1个单倍型。
2.2 单倍型及进化关系
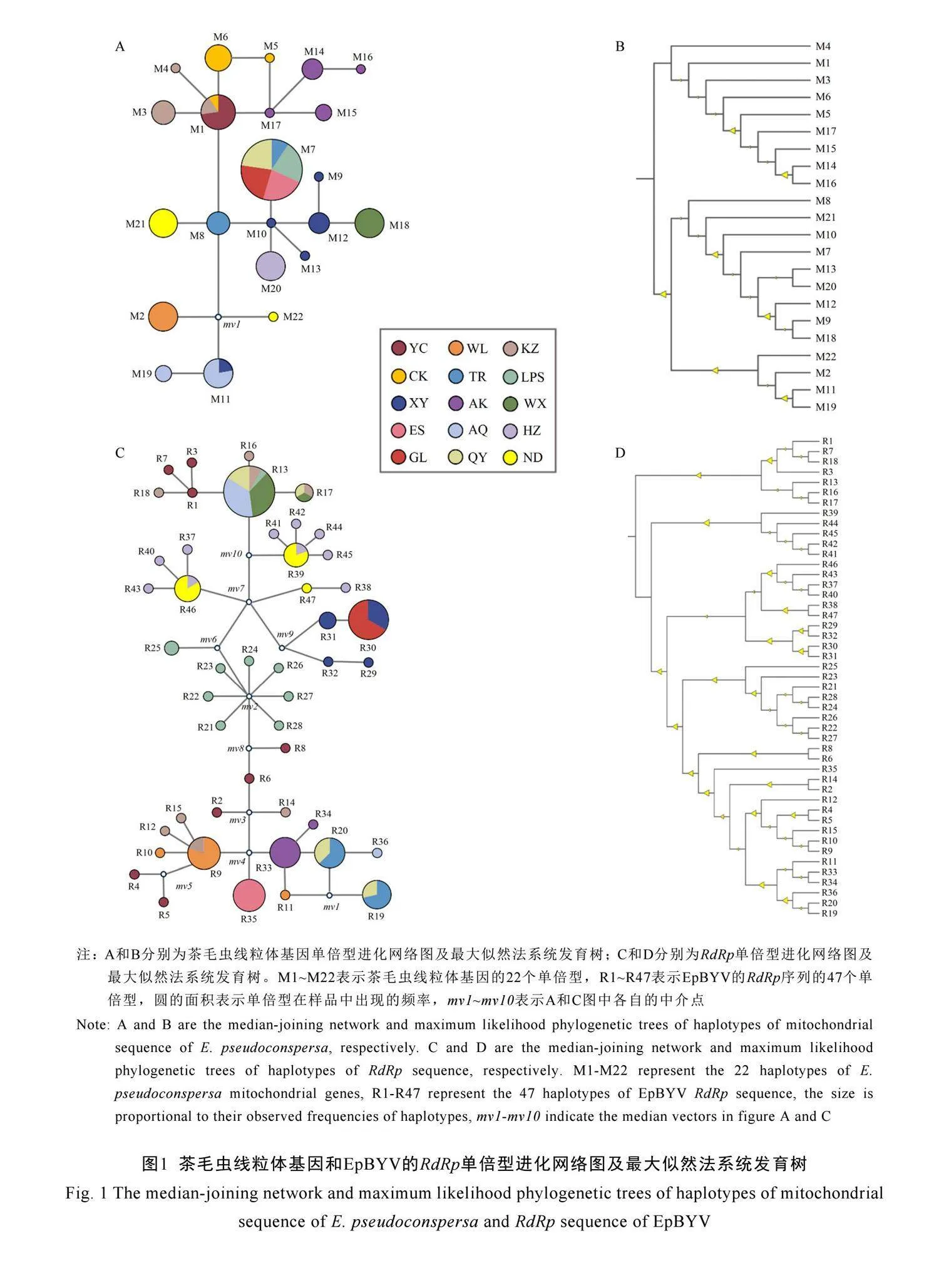
在茶毛虫线粒体基因序列的22个单倍型中,种群间共享的单倍型有3个,占比较少(13.64%),表明茶毛虫线粒体基因序列在种群间的遗传分化程度较高。共享单倍型中M7的出现频率最高(44次),占所有个体的29.73%,其次是M1(11次)和M11(9次)。从种群来看,永川、武隆、六盘水、无锡、恩施、杭州、桂林、清远等地的8个种群内部均无突变位点,各自仅有1种单倍型,其中六盘水、恩施、桂林和清远4个地区的种群共享单倍型M7,此外信阳种群有5个单倍型,安康种群4个,遗传分化程度相对较大。单倍型的进化网络图显示,大部分来自相同种群的单倍型聚在一起(开州、城口、安康、信阳、安庆)。进化网络图在结构上呈现明显的两个分枝(图1A),上部一枝有9个单倍型,包含永川、开州、城口、安康4个种群的38个样本;下部一枝有13个单倍型,由其他11个种群110个样本组成。安康在传统茶区划分中被分在江北茶区,但在地理位置上与上部一枝上的开州、城口共同位于秦巴山区,而武隆虽隶属于重庆市,但在地理位置上与恩施、铜仁共同位于武陵山区,表明茶毛虫线粒体基因序列的变异与种群的地理位置有一定的关系。采用最大似然法构建22个单倍型的系统发育树,显示其分枝结构与网络进化图相吻合,进化网络图中的上下两枝在进化树均形成单系群,并互为姊妹群(图1B)。
不同种群EpBYV的RdRp序列的47个单倍型中有7个共享单倍型,占比较小(14.89%)。共享单倍型中R13出现的频率最高(25次),占所有个体的18.12%,其次是R30(15次)。种群方面,桂林种群仅有1个单倍型,其他种群的单倍型数在2~10个,其中杭州和永川种群均是1个样本为1个单倍型,与之对应的是两个种群的茶毛虫线粒体基因序列在种群内部无突变位点,由此可见EpBYV的RdRp序列比茶毛虫线粒体序列的突变速率更快,种群内遗传分化更大。RdRp序列的单倍型数量较大,相对于茶毛虫线粒体序列单倍型的进化网络图,其进化网络图结构更为复杂,难以通过地理种群明确划分枝系。大部分来自相同种群的单倍型较好地聚在一起,但永川、开州、安庆、六盘水种群的单倍型在图中分布距离较远(图1C),表明这4个种群内部出现较大的遗传分化。此外,地理种群间的进化关系也更为复杂,在图1A中,茶毛虫的武隆种群与重庆的其他3个种群的进化距离较远,在图1C中,EpBYV的永川、开州种群的部分单倍型与武隆种群距离较远,但另一些单倍型与武隆种群聚在一起(R4、R5、R9、R10、R12、R15)。47个单倍型的最大似然法系统发育树与网络进化图结构相似,在进化图上聚在一起的单倍型也以较高的支持率在进化树上聚为一枝(图1D)。
2.3 种群遗传结构分析
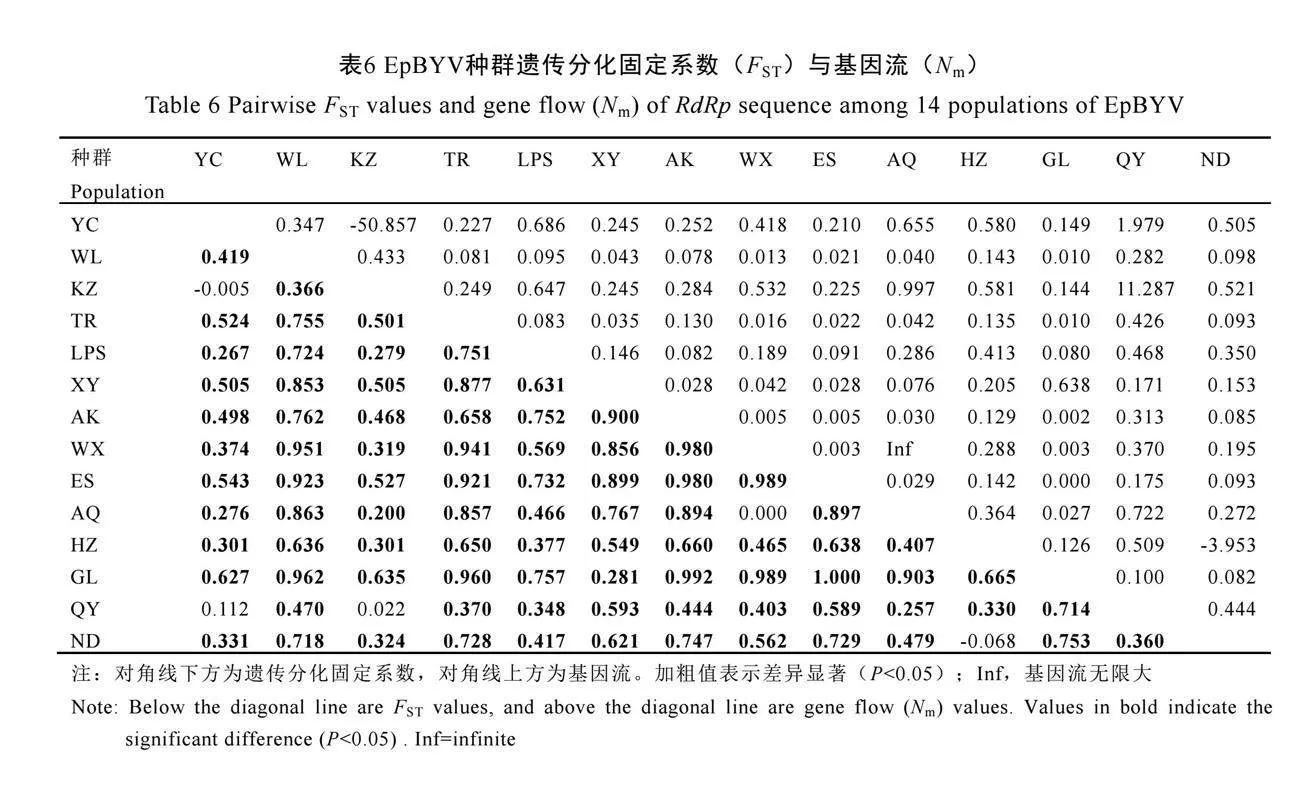
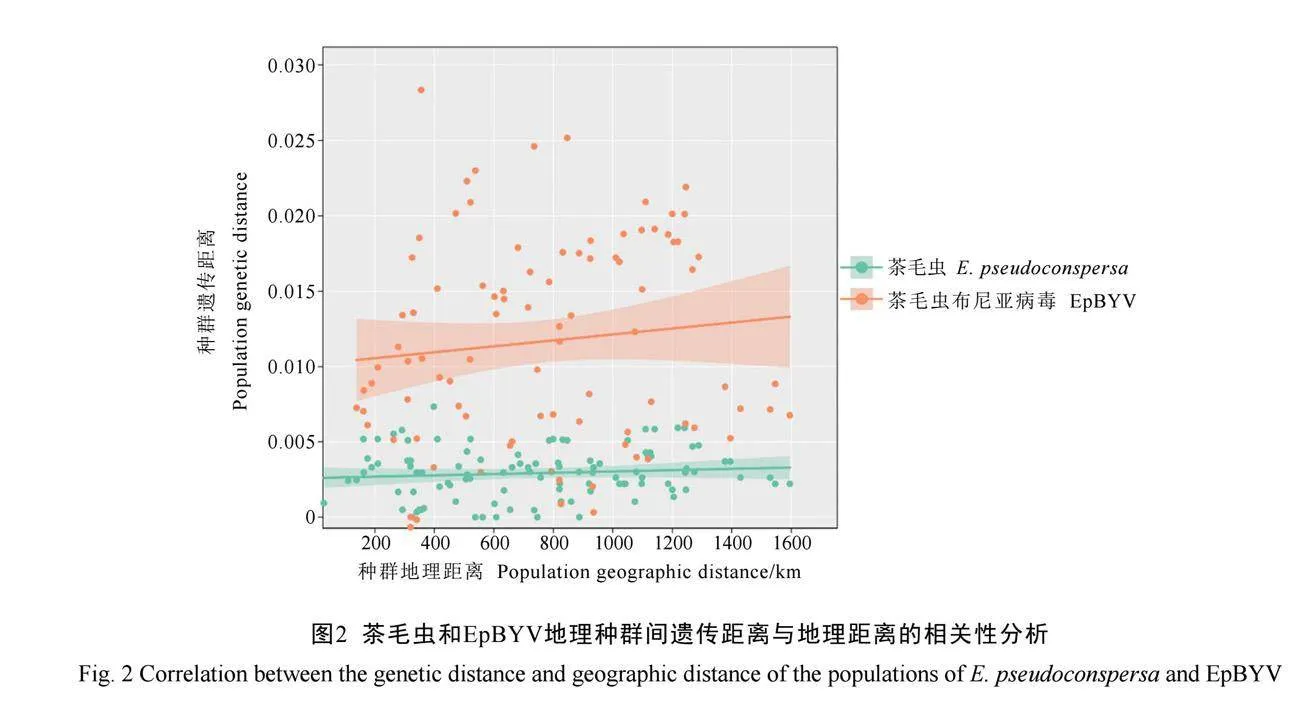
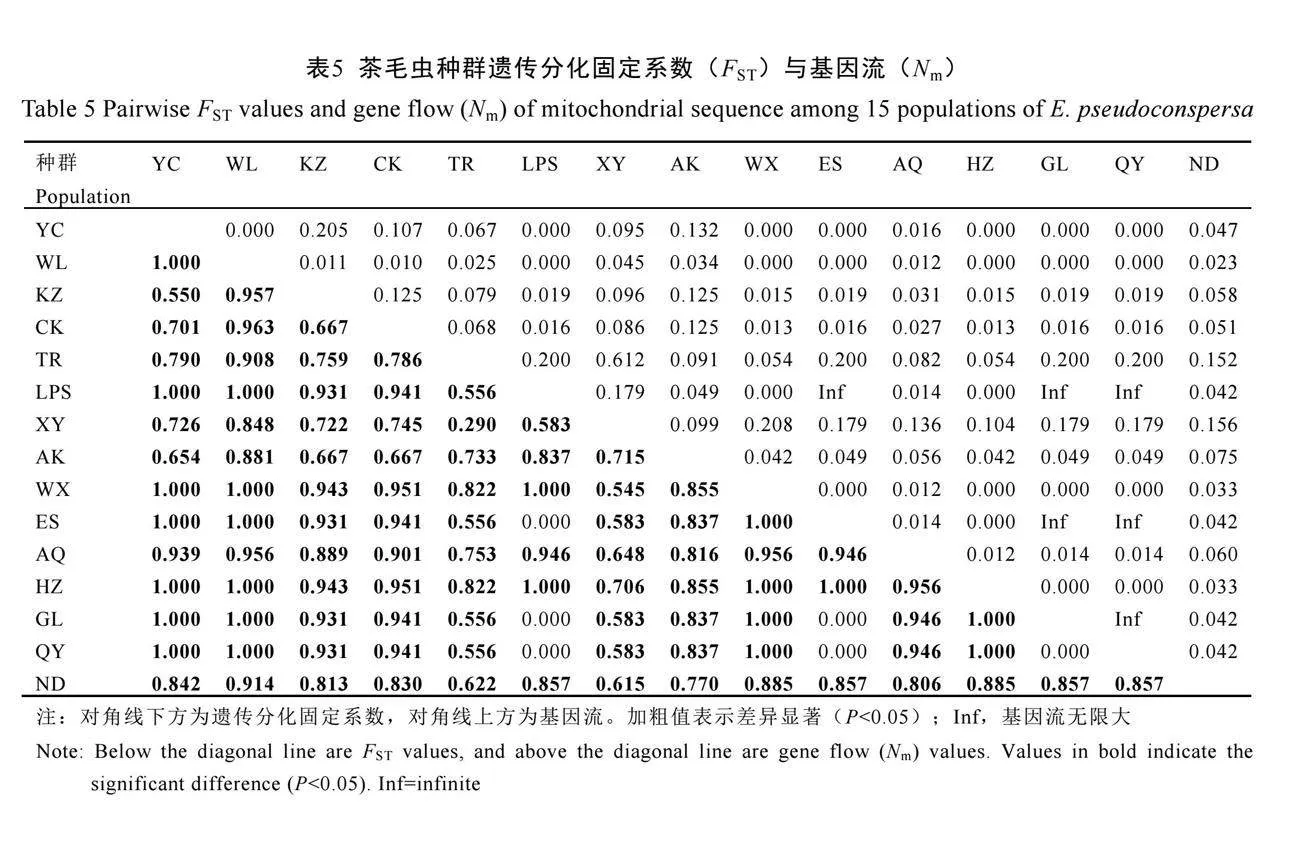
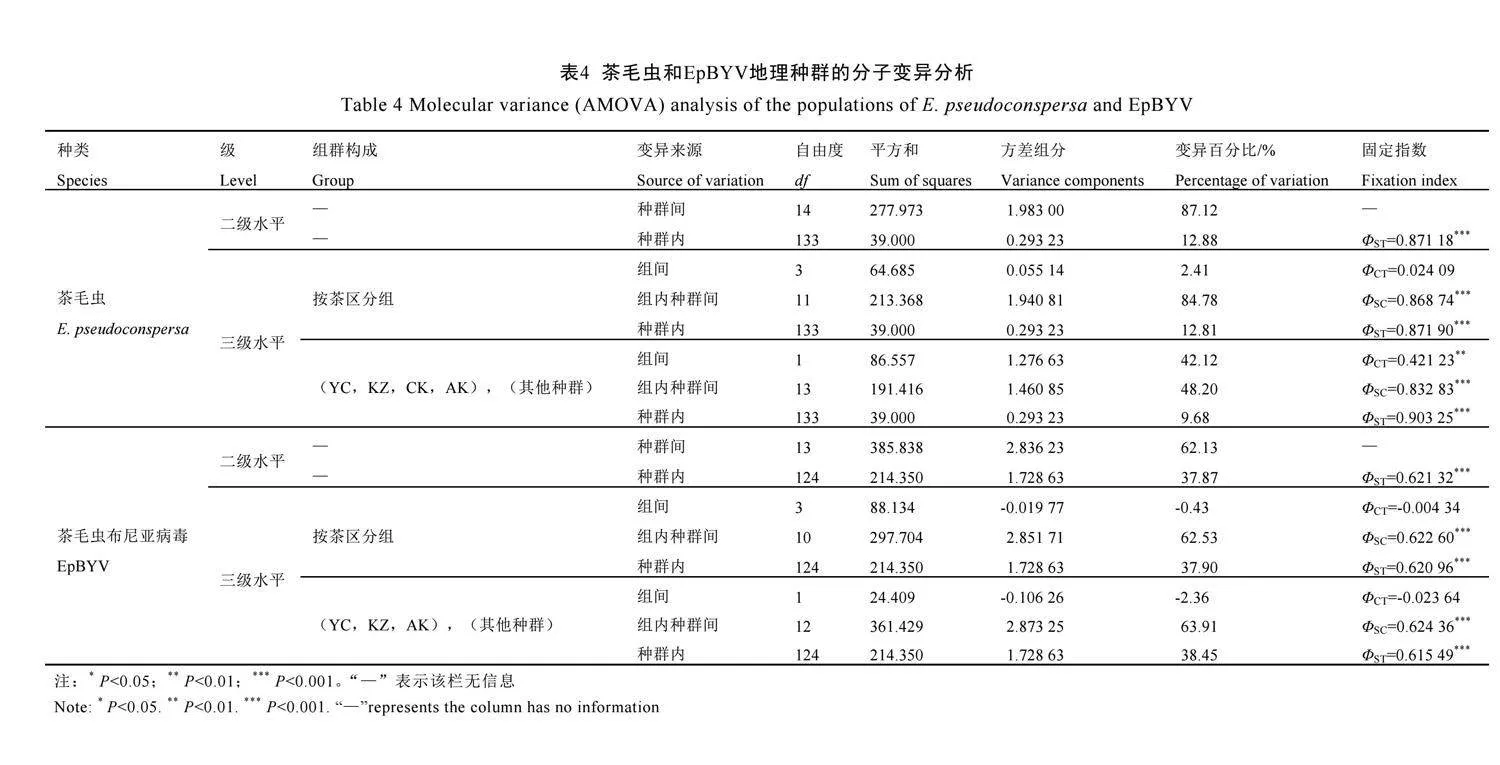
多个水平的AMOVA分析均显示茶毛虫存在显著的遗传分化(表4),两级水平分析显示茶毛虫具有显著的遗传结构(ΦST=0.871 18,P<0.001),其中87.12%的变异来自于种群之间,仅12.88%的变异来自于种群内部。105个种群对间的遗传分化指数FST值有99个达到显著水平,数值为0.290~1.000(表5),支持茶毛虫多数地理种群间存在显著的遗传分化,与之对应的是多数种群对间基因流Nm水平较低(表5)。三级水平AMOVA分析将地理种群按传统茶区(表1)分成4个组时,茶毛虫不具有显著的遗传结构(ΦCT=0.024 09,P>0.05),遗传变异主要来自于组内种群间,即各地理种群之间的差异;但根据进化网络结构图中上下两枝分为2个组时,茶毛虫存在显著的遗传结构(ΦCT=0.421 23,P<0.01),茶毛虫在两个组之间显著分化,组间变异占42.12%。对EpBYV二级水平AMOVA分析显示其遗传结构显著(ΦST=0.621 32,P<0.001),遗传变异大部分来自于种群之间(62.13%),种群内部的遗传变异较小(37.87%),但仍大于茶毛虫各种群内部的遗传变异占比(12.88%)。与茶毛虫类似,EpBYV种群对间的遗传分化指数FST值大多数达到了显著水平(表6),Nm值大多数小于1(表6),支持EpBYV的地理种群间具有显著的遗传分化,基因交流较少。三级水平AMOVA分析显示,EpBYV无显著的遗传结构,两种分组下ΦCT的P值均大于0.05,组间的遗传变异占比均为负值。种群间遗传距离与地理距离的相关性分析结果显示,茶毛虫(R2=0.010,P=0.305)和EpBYV(R2=0.012,P=0.296)的遗传距离与地理距离的相关性不显著(图2),且多数EpBYV种群间的遗传距离较茶毛虫种群间更大。进一步计算发现,EpBYV种群间的遗传距离与茶毛虫对应种群
间遗传距离的相关性不显著(R2=0.006,P=0.461),表明EpBYV进化方向不受茶毛虫的影响。
2.4 种群历史动态分析
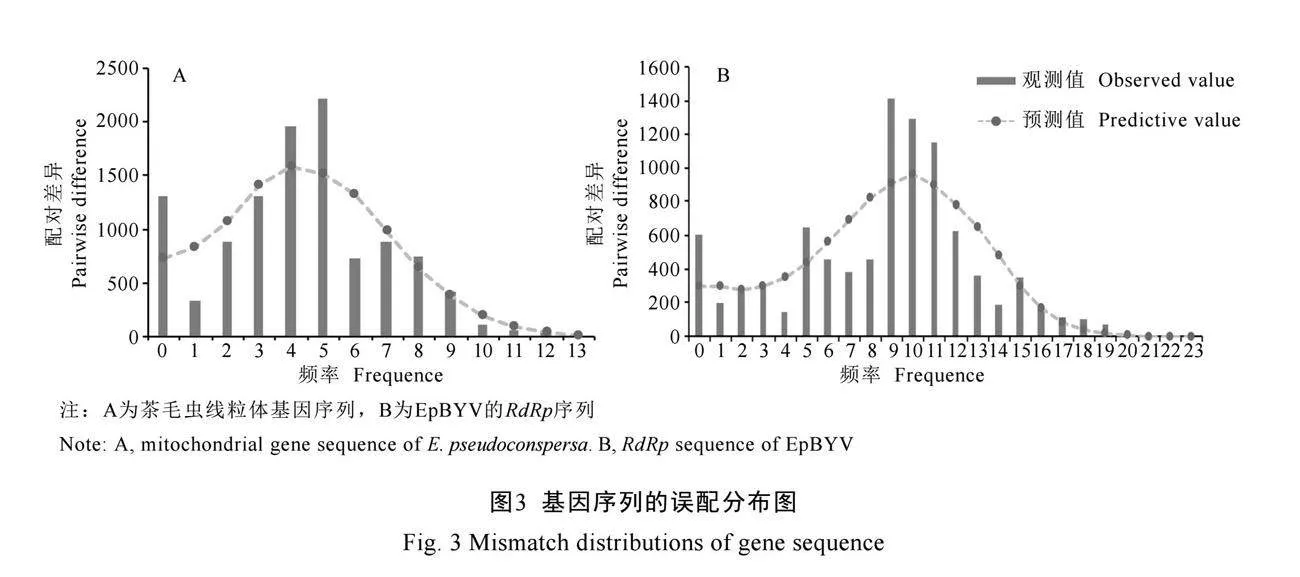
采用中性检验和误配分析进行茶毛虫和EpBYV历史动态分析。茶毛虫7个有多态性位点的种群中,城口和宁德种群的Tajima’s D值显著小于0,可能是种群扩张引起种群偏离了遗传平衡状态,但Fu’s Fs值未达到显著水平;其他种群的中性检验值均不显著,说明群体处于较稳定状态;总群体的中性检验虽均为负值,但未达到显著水平(表3)。茶毛虫总群体的误配分布表现为单峰(图3A),且误配分布统计参数粗糙度指数(Raggedness index,rg)为0.036 97,期望与观测错配分布之间的平方和(SSD)为0.013 21,两个值均较小,且未达到显著水平,推测我国茶毛虫群体可能经历了较弱的突然增长过程。EpBYV地理种群的中性检验显示,六盘水种群的Tajima’s D值和Fu’s Fs值均显著小于0,武隆、安庆种群的Tajima’s D值和杭州种群的Fu’s Fs值均显著小于0,推测这4个种群可能经历了不同程度扩张。EpBYV总群体的Tajima’s D值和Fu’s Fs值均显著小于0,其中Fu’s Fs值达到了极显著水平,且误配分布图表现为明显的单峰(图3B),rg值为0.020 78(P=0.01),SSD值为0.011 51(P=0.07),表明EpBYV可能经历了突然增长过程。
3 讨论
本研究采集并提取了茶毛虫的DNA和RNA,以此为模板分别扩增茶毛虫的线粒体COI、ND5和EpBYV的RdRp编码序列,其中茶毛虫线粒体两个基因的扩增及比对成功率均为100%,表明所采集样本均为茶毛虫;而EpBYV的RdRp序列扩增成功率为93.24%,在城口种群10个样本中未能成功扩增,原因可能是茶毛虫城口种群未感染EpBYV或体内病毒量极低。因此,148个茶毛虫样本的EpBYV感染率大于或等于93.24%,表明该病毒广泛分布于茶毛虫的田间种群中。
基于线粒体COI和ND5合并序列分析茶毛虫的种群遗传结构,二级水平的AMOVA分析结果和遗传分化指数ΦST值均支持茶毛虫的遗
传变异主要来自于地理种群之间,种群之间遗传分化极显著。三级水平AMOVA分析显示,我国传统茶区划分不能指示茶毛虫地理种群间的遗传分化程度,参考进化网络结构图能较为正确地指示种群间的分化程度。将该分组置于我国地势中可以发现,图1A中上部一枝的4个地理种群(永川、开州、城口、安康)位于我国地势的第二阶梯,而下部一枝的地理种群均位于第三阶梯及第二、三阶梯的分界上,推测茶毛虫在两个区域的显著分化可能是两个阶梯海拔和气候差异引起的。突变、自然选择、基因流和随机遗传漂变(Random genetic drift)被公认为是物种进化的四大动力[29]。其中,突变、自然选择和随机遗传漂变加大种群间的遗传分化,基因流则可以阻止种群间的分化,因此茶毛虫种群间低水平的基因流与高水平的遗传分化相吻合,由此推测茶毛虫扩散能力不强。同样不具备长距离传播能力的茶小绿叶蝉、茶网蝽和茶棍蓟马均显示出较高的遗传分化[22-24]。地理种群间的显著遗传分化也可能是造成不同地理种群茶毛虫对同株EpNPV敏感性不同的原因。基于RdRp编码序列分析EpBYV的种群遗传结构,ΦST值显示EpBYV种群之间遗传分化显著,二级水平AMOVA分析支持遗传变异主要来自于地理种群之间,但种群内部的遗传变异占比(37.87%)明显大于茶毛虫种群内部的遗传变异占比(12.88%),遗传多样性参数(h=0.935 26、π=0.017 95)也支持EpBYV的种群具有更大的遗传变异(表3),表明EpBYV与茶毛虫的进化差异明显,这与茶毛虫成虫飞翔能力较弱,对病毒水平传播不足相关。病毒毒株的遗传分化可能影响其对宿主的毒力水平[8],EpBYV的显著遗传分化和快速突变有利于进化出对茶毛虫产生致病力的毒株。
本研究的种群历史动态分析采用中性检验和误配分析相结合,当中性检验Tajima’s D和Fu’s Fs值均为显著负值时,说明所研究的种群可能经历了突然扩张事件[32],误配分布表现为单峰表示种群在近期出现过群体扩张事件或持续增长模式[36-37]。茶毛虫地理种群中,城口和宁德种群的Tajima’s D值显著小于0,推测近期经历了群体扩张事件。茶毛虫总群体的中性检验均为不显著的负值,误配分布表现为单峰,推测茶毛虫总群体可能经历了较弱的群体扩张。EpBYV总群体的Tajima’s D值和Fu’s Fs值均显著小于0,其中Fu’s Fs值达到了极显著水平,误配分布图为明显的单峰,表明EpBYV群体可能经历了突然增长过程,近似星状分布的单倍型进化网络图也佐证EpBYV在近期经历了种群有效数量突然增长的过程。此外,RdRp序列具有较高水平的单倍型多样性(0.935 26)和相对较低的核苷酸多样性(0.017 95),也支持EpBYV种群经历了种群扩张事件[38]。EpBYV群体扩张事件主要出现在六盘水、武隆、安庆、杭州4个地理种群中。
综上所述,采自我国四大茶区15个地区的148个样本均确认为茶毛虫,且其群体相对稳定,近期可能仅经历了较弱的群体扩张事件,仅城口和宁德种群经历了较明显的群体扩张,表明茶毛虫在部分茶区的适宜环境条件下也具有潜在暴发的风险;在茶毛虫总群体相对稳定的背景下,EpBYV种群经历了明显的种群扩张事件,说明EpBYV即使在经历种群萎缩后,仍可以迅速由少量个体建立庞大的种群。同时,EpBYV的种群扩张将增加病毒在复制时的突变机率,这有利于进化出对茶毛虫产生致病力的毒株。EpBYV在茶毛虫群体中的感染率和种群扩张能力均较高,用于茶毛虫生物防治具有较好的潜力。
参考文献
[1] 张汉鹄, 谭济才. 中国茶树害虫及其无公害治理[M]. 合肥: 安徽科学技术出版社, 2004.
Zhang H H, Tan J C. Chinese tea pests and their pollution-free control [M]. Hefei: Anhui Science and Technology Press, 2004.
[2] 戈峰, 王永模, 刘向辉. 茶毛虫性引诱剂诱杀效果分析[J]. 昆虫知识, 2003, 40(3): 237-239.
Ge F, Wang Y M, Liu X H. The effectiveness of synthetic sex pheromone by mass trapping as a control method for the tea tussock moth, Euproctis pseudoconspersa [J]. Entomological Knowledge, 2003, 40(3): 237-239.
[3] 韩方岸, 胡云, 李世荣, 等. 茶毛虫生态特性及人群皮炎暴发的观察[J]. 中国寄生虫病防治杂志, 2005, 18(5): 378-381.
Han F A, Hu Y, Li S R, et al. Observation on the ecology of tea Euproctis pseudoconspersa strand and the outbreak of larvae dermatitis in crowd [J]. Chinese Journal of Parasitic Disease Control, 2005, 18(5): 378-381.
[4] Dong W W, Dong S Y, Jiang G F, et al. Characterization of the complete mitochondrial genome of tea tussock moth, Euproctis pseudoconspersa (Lepidoptera: Lymantriidae) and its phylogenetic implications [J]. Gene, 2016, 577(1): 37-46.
[5] 彭萍, 王晓庆, 李品武. 茶树病虫害测报与防治技术[M]. 北京: 中国农业出版社, 2013.
Peng P, Wang X Q, Li P W. Forecast and control techniques of tea diseases and pests [M]. Beijing: China Agriculture Press, 2013.
[6] Hazarika L K, Bhuyan M, Hazarika B N. Insect pests of tea and their management [J]. Annual Review of Entomology, 2009, 54: 267-284.
[7] Ye G Y, Xiao Q, Chen M, et al. Tea: biological control of insect and mite pests in China [J]. Biological Control, 2014, 68: 73-91.
[8] 付建玉, 席羽, 唐美君, 等. 茶毛虫核型多角体病毒不同分离株的毒力水平与遗传结构关系分析[J]. 茶叶科学, 2011, 31(4): 289-294.
Fu J Y, Xi Y, Tang M J, et al. Study on the relationship between virulence and genetic structure of four wild isolates of Euproctis pseudoconspersa nuclear polyhedrosis virus [J]. Journal of Tea Science, 2011, 31(4): 289-294.
[9] 厉晓腊, 金轶伟, 柴一秋, 等. 茶毛虫核型多角体病毒对茶毛虫的致病性研究[J]. 茶叶科学, 2006, 26(4): 265-269.
Li X L, Jin Y W, Chai Y Q, et al. The pathogenicity of Euproctis pseudoconspersa nuclear polyhedrosis virus on the larvae of tea tussork (Euproctis pseudoconspersa Strand) [J]. Journal of Tea Science, 2006, 26(4): 265-269.
[10] 王礼中, 肖强, 张传溪. 茶毛虫核型多角体病毒ph基因抗体的制备与利用[J]. 茶叶科学, 2008, 28(4): 260-266.
Wang L Z, Xiao Q, Zhang C X. Preparation of the antibody against EupsNPV polyhedrin and its utilization in detection of the viral pesticide [J]. Journal of Tea Science, 2008, 28(4): 260-266.
[11] 李兆群. 我国茶园有害生物生物防治技术研究及应用[J]. 中国茶叶, 2022, 44(5): 8-12.
Li Z Q. Research and application of tea pest control technology in China [J]. China Tea, 2022, 44(5): 8-12.
[12] 陈宗懋, 蔡晓明, 周利, 等. 中国茶园有害生物防控40年[J]. 中国茶叶, 2020, 42(1): 1-8.
Chen Z M, Cai X M, Zhou L, et al. Developments on tea plant pest control in past 40 years in China [J]. China Tea, 2020, 42(1): 1-8.
[13] 冷杨, 肖强, 殷坤山. 茶毛虫核型多角体病毒Bt混剂的作用特性[J]. 植物保护学报, 2007, 34(2): 177-181.
Leng Y, Xiao Q, Yin K S. The effect specialty of EpNPV-Bt preparation [J]. Acta Phytophylacica Sinica, 2007, 34(2): 177-181.
[14] 唐美君, 殷坤山, 郭华伟, 等. 茶毛虫核型多角体病毒和Bt混剂的配比筛选及药效评价[J]. 植物保护, 2010, 36(5): 165-167.
Tang M J, Yin K S, Guo H W, et al. Screening for ratio of EpNPV mixed with Bt and evaluation of its control effect [J]. Plant Protection, 2010, 36(5): 165-167.
[15] 王晓庆. 茶毛虫的发生规律及其感染的病毒分析[D]. 重庆: 西南大学, 2021.
Wang X Q. Study on the occurrence and viral infection of Euproctis pseudoconspersa [D]. Chongqing: Southwest University, 2021.
[16] Wang X Q, Gu Q Y, Zhang W, et al. Prevalence of a novel bunyavirus in tea tussock moth Euproctis pseudoconspersa (Lepidoptera: Lymantriidae) [J]. Journal of Insect Science, 2021, 21(4): 5. doi: 10.1093/jisesa/ieab045.
[17] Marklewitz M, Zirkel F, Kurth A, et al. Evolutionary and phenotypic analysis of live virus isolates suggests arthropod origin of a pathogenic RNA virus family [J]. PNAS, 2015, 112(24): 7536-7541.
[18] Bolling B G, Olea-Popelka F J, Eisen L, et al. Transmission dynamics of an insect-specific flavivirus in a naturally infected Culex pipiens laboratory colony and effects of co-infection on vector competence for West Nile virus [J]. Virology, 2012, 427(2): 90-97.
[19] Newman C M, Cerutti F, Anderson T K, et al. Culex flavivirus and West Nile virus mosquito coinfection and positive ecological association in Chicago, United States [J]. Vector-Borne and Zoonotic Diseases, 2011, 11(8): 1099-1105.
[20] 王星驰, 晏嫦妤. 茶毛虫发生规律与生物防治研究进展[J]. 广东茶业, 2023(5): 16-22.
Wang X C, Yan C Y. Research progress on occurrence regularity and biological control of Euproctis pseudoconspersa [J]. Guangdong Tea Industry, 2023(5): 16-22.
[21] 马蕊, 白阿先, 卢木朴. 4种生物农药防治云南绿春县茶毛虫的效果研究[J]. 昆明学院学报, 2022, 44(3): 53-56.
Ma R, Bai A X, Lu M P. Effect of four kinds of biological pesticides on controlling Euproctis pseudoconspersa in Lüchun county of Yunnan province [J]. Journal of Kunming University, 2022, 44(3): 53-56.
[22] 李金玉, 牛东升, 陈杰, 等. 茶园周边景观格局对茶小绿叶蝉种群遗传结构的影响[J]. 昆虫学报, 2020, 63(10): 1242-1259.
Li J Y, Niu D S, Chen J, et al. Effects of landscape pattern around tea plantation on the population genetic structure of the tea green leafhopper, Empoasca onukii (Hemiptera: Cicadellidae) [J]. Acta Entomologica Sinica, 2020, 63(10): 1242-1259.
[23] 陈世春, 江宏燕, 廖姝然, 等. 基于COI基因解析我国茶网蝽种群遗传多样性和遗传结构[J]. 茶叶科学, 2023, 43(6): 795-805.
Chen S C, Jiang H Y, Liao S R, et al. Analysis of genetic diversity and genetic structure in geographic populations of Stephanitis chinensis from China based on mitochondrial DNA COI sequence [J]. Journal of Tea Science, 2023, 43(6): 795-805.
[24] 罗林丽, 孟泽洪, 李帅, 等. 中国南方茶棍蓟马地理种群遗传分化分析[J]. 昆虫学报, 2022, 65(4): 500-511.
Luo L L, Meng Z H, Li S, et al. Genetic differentiation analysis of geographical populations of Dendrothrips minowai (Thysanoptera: Thripidae) in South China [J]. Acta Entomologica Sinica, 2022, 65(4): 500-511.
[25] Zhang D X, Hewitt G M. Assessment of the universality and utility of a set of conserved mitochondrial COI primers in insects [J]. Insect Molecular Biology, 1997, 6(2): 143-150.
[26] Simon C, Buckley T R, Frati F, et al. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA [J]. Annual Review of Ecology Evolution and Systematics, 2006, 37: 545-579.
[27] Rozas J. DNA sequence polymorphism analysis using DnaSP [J]. Methods in Molecular Biology, 2009, 537: 337-350.
[28] Rozas J, Ferrer-Mata A, Sanchez-DelBarrio J C, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets [J]. Molecular Biology and Evolution, 2017, 34(12): 3299-3302.
[29] 魏丹丹. 书虱种群遗传多样性及线粒体基因组进化研究[D]. 重庆: 西南大学, 2012.
Wei D D. Population genetic diversity and mitochondrial genome analysis of Psocids (Psocoptera: Liposcelididae) [D]. Chongqing: Southwest University, 2012.
[30] Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis [J]. Evolutionary Bioinformatics Online, 2005, 1: 47-50.
[31] Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism [J]. Genetics, 1989, 123(3): 585-595.
[32] Fu Y X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection [J]. Genetics, 1997, 147(2): 915-925.
[33] Chen J, Liu Q, Gao L W. Visual tea leaf disease recognition using a convolutional neural network model [J]. Symmetry, 2019, 11(3): 343. doi: 10.3390/sym11030343.
[34] Bandelt H J, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies [J]. Molecular Biology and Evolution, 1999, 16(1): 37-48.
[35] Guindon S, Dufayard J F, Lefort V, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0 [J]. Systematic Biology, 2010, 59(3): 307-321.
[36] Wei D D, Yuan M L, Wang B J, et al. Population genetics of two asexually and sexually reproducing psocids species inferred by the analysis of mitochondrial and nuclear DNA sequences [J]. Plos One, 2012, 7(3): e33883. doi: 10.1371/journal.pone.0033883.
[37] Rogers A R, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences [J]. Molecuar Biology and Evolution, 1992, 9(3): 552-569.
[38] Slatkin M. Isolation by distance in equilibrium and non-equilibrium populations [J]. Evolution, 1993, 47(1): 264-279.
