
保种场涪陵水牛及西南地区水牛品种间遗传结构与ROH分析
2024-09-19 00:00:00屠芸曾雅楠张蒸豪洪瑞王震吴平周泽洋叶艺茹杜亚楠左福元张龚炜
畜牧兽医学报 2024年5期




摘 要: 旨在对保种场涪陵水牛及西南地区其他沼泽型水牛品种进行群体遗传结构和连续纯合片段(ROH)分析,为涪陵水牛保种与遗传改良提供理论依据。本研究对保种场26头涪陵水牛以及1头杂交水牛(FM_HY)进行全基因组重测序,从NCBI下载了68头西南地区沼泽型水牛和7头河流型水牛的基因组重测序数据,基于所有的基因组数据获得的高质量单核苷酸多态性(SNPs)位点进行遗传结构、ROH以及近交系数的分析。全基因组重测序结果显示,涪陵水牛平均测序深度为10×,质量控制后共鉴定出14 326 437个SNPs。群体遗传结构分析显示,保种场内涪陵水牛与毗邻区域的宜宾水牛、贵州水牛、贵州白水牛、盐津水牛间具有明显的遗传结构差异,德宏水牛和滇东南水牛与上述5个品种区别均明显;当分群数量为K=6时,涪陵水牛可划分为2个类群,与毗邻区域水牛品种相比具有一类独特的群体结构。涪陵水牛、宜宾水牛、贵州水牛这3个品种间共检测到2 722个ROHs,其中涪陵水牛检测到1 593个ROHs,且1~2 Mb的短ROH片段占比达78.15%;仅在涪陵水牛群体中检测到8个大于16 Mb的长ROH片段,最长为24.56 Mb。基于ROH计算的保种场涪陵水牛平均近交系数为0.056 8,其中7头个体近交系数较高(0.062 5~0.125 0);贵州水牛和宜宾水牛的平均近交系数分别为0.034 6和0.041 0。综上,涪陵水牛保种群可分为2个类群,与毗邻区域水牛品种相比具有一类独特的群体结构;保种场内7头涪陵水牛个体存在近交风险,迫切需要加强涪陵水牛的资源保护与利用工作。
关键词: 涪陵水牛;群体遗传结构;SNP;ROH;近交系数
中图分类号: S823.83
文献标志码:A""" 文章编号:0366-6964(2024)05-1989-10
收稿日期:2023-11-20
基金项目:重庆市技术创新与应用发展专项重点项目(cstc2021jscx-gksbX0012);重庆市农业种质资源精准鉴定项目(23316)
作者简介:屠 芸(1997-),女,江苏连云港人,硕士生,主要从事动物遗传育种研究,E-mail: yuntu01@163.com
*通信作者:张龚炜,主要从事动物遗传育种研究,E-mail: zgw-vip@163.com ;左福元,主要从事牛的遗传改良及反刍动物营养研究,E-mail: zfuyuan@163.com
Genetic Structure and Runs of Homozygosity Analysis of Fuling Buffalo and Southwest Buffalo
Breeds
TU" Yun1,2, ZENG" Yanan1,2, ZHANG" Zhenghao1,2, HONG" Rui1,2, WANG" Zhen3, WU" Ping4,
ZHOU" Zeyang1,2, YE" Yiru1,2, DU" Yanan1,2, ZUO" Fuyuan1,2*, ZHANG" Gongwei1,2*
(1.College of Animal Science and Technology, Southwest University, Rongchang 402460," China;
2.Beef Cattle Engineering and Technology Research Center of Chongqing, Rongchang 402460," China;
3.Chongqing Animal Husbandry Technology Popularization Station, Chongqing 401121," China;
4.Chongqing Nanchuan Animal Husbandry, Veterinary and Fishery Center, Nanchuan 408400," China)
Abstract:" This study aimed to analyze the population genetic structure and runs of homozygosity (ROH) of Fuling buffalo and other swamp buffalo breeds in southwest China, and provide the theoretical basis for conservation and genetic improvement of Fuling buffalo. In this study, 26 Fuling buffaloes and one hybrid buffalo (FM_HY) were sequenced using whole-genome resequencing technology. The genome resequencing data of 68 swamp buffalo individuals in southwest China and 7 river buffalo individuals were downloaded from the NCBI database. All the genome data were used to obtain high-quality single nucleotide polymorphisms (SNPs) and to analyze the population genetic structure, ROH and inbreeding coefficient. The whole-genome resequencing results showed that the average sequencing depth of Fuling buffalo was 10×, and a total of 14 326 437 SNPs were identified after quality control. The population genetic structure analysis showed that there were significant differences between the Fuling buffalo and the neighboring areas swamp buffalo breeds including Yibin buffalo, Guizhou buffalo, Guizhou White buffalo and Yanjin buffalo. Dehong buffalo and Diandongnan buffalo were significantly different from the above 5 breeds. When the number of clusters was K=6, Fuling buffalo was divided into two groups, which had a unique population structure compared to the neighboring areas buffalo breeds. A total of 2 722 ROHs were detected in Fuling buffalo, Yibin buffalo and Guizhou buffalo. The 1 593 ROHs were identified in Fuling buffalo, and the 78.15% proportion of ROH was the short ROH with 1-2 Mb. Eight long ROH fragmentsgt;16 Mb were only detected in Fuling buffalo population, the longest being 24.56 Mb. Based on ROH, the average inbreeding coefficient of Fuling buffalo was 0.056 8, of which 7 individuals had higher inbreeding coefficient in 0.062 5-0.125 0. The average inbreeding coefficients of Guizhou buffalo and Yibin buffalo were 0.034 6 and 0.041 0, respectively. In conclusion, the population of Fuling buffalo in the conservation farm can be divided into two groups, which have a unique population structure compared to the other neighboring areas buffalo breeds, and 7 Fuling buffalo individuals in the conservation farm have inbreeding risk, so it is urgent to strengthen the protection and utilization of Fuling buffalo resources.
Key words: Fuling buffalo; population genetic structure; SNP; ROH; inbreeding coefficient
*Corresponding authors:ZHANG Gongwei, E-mail: zgw-vip@163.com; ZUO Fuyuan, E-mail: zfuyuan@163.com
涪陵水牛(Fuling buffalo,FL)是我国优良的沼泽型地方水牛品种(2n=48),具有体躯结实、结构紧凑、抗病力强、适应性好、耐粗饲、耐高温高湿、役力强、易于饲养和管理等特点,已被列入《国家畜禽遗传资源品种名录(2020年版)》[1]。根据《重庆市畜禽遗传资源志》[2]记载,2012年涪陵区、南川区、綦江区3个主产区县共存栏涪陵水牛6.8万头,而2022年第三次全国畜禽遗传资源普查发现全重庆市仅有5 990头。重庆市在南川区建有涪陵水牛保种场,但场内牛群的遗传结构及遗传多样性缺乏科学数据支撑,不利于涪陵水牛保种工作的开展。
1964年,涪陵水牛开展了本品种的选种选配,其体型和役用性能均有所提高[3];1974年从广西引进摩拉公水牛开展杂交改良[4],产奶性能得到提升。为了解涪陵水牛的种质特性,早期学者对涪陵水牛的染色体结构、蛋白质多态性、mtDNA D-loop序列等方面进行研究[5-10]。结果表明,涪陵水牛的染色体数目、臂数、类型及性染色体大小等均与其他文献介绍的沼泽型水牛相同[5],并且与位于长江流域的德昌水牛、江汉水牛、滨湖水牛和海子水牛间的遗传差异较小,亲缘关系较近[6-7];淀粉酶多态性分析发现,涪陵水牛与德宏水牛间的遗传距离较大[8]。mtDNA D-loop区序列分析认为中国地方水牛可能存在两大母系起源[9]。谢文美等[10]利用mtDNA D-loop序列分析发现涪陵水牛较其他沼泽型和江河型水牛的遗传多样性更为丰富,且具有2个母系起源。二代高通量测序技术可在全基因组范围检测单核苷酸多态性(single nucleotide polymorphism,SNP)位点,并被广泛用于畜禽动物种群遗传多样性、群体结构和亲缘关系分析[11-13]。连续纯合片段(ROH)被定义为基因组中长而连续的纯合子延伸,其纯合子片段长度足够长,它是由同一祖先遗传下来的两种完全相同的单倍型所构成,能够用来评估家畜近交系数、推断种群历史、辅助解析某些复杂性状的形成机制[14-17]。连续性纯合片段(runs of homozygosity, ROH)是基因组中的一段长纯合片段,不同长度的ROH和个体与共同祖先的亲缘背景相关联,能够用来评估家畜近交系数、推断种群历史、辅助解析某些复杂性状的形成机制[14-17]。
为此,本研究利用全基因组重测序技术基于SNP数据对保种场涪陵水牛及西南地区其他水牛品种进行遗传结构和ROH分析,将为涪陵水牛保种与遗传改良提供理论依据。
1 材料与方法
1.1 试验设计与试验动物
本研究核心目的是了解保种场内涪陵水牛与毗邻区域水牛品种间遗传结构的差异以及保种场内涪陵水牛的近交水平。为此:1)采集保种场内21头成年水牛(FL1~FL21),用于遗传结构和基于ROH近交系数分析;2)采集本场5头自群繁育的犊牛(FL22~26),主要目的是进行系谱纠偏;3)采集1头杂交母水牛(FM_HY)(摩拉水牛()×涪陵水牛(♀)的后代),主要是为排除NCBI下载数据具有河流型与沼泽型水牛的杂交样本。采集上述27头水牛耳组织保存于装有1 mL 75%酒精的1.5 mL离心管中,于-20 ℃保存。从NCBI下载11个水牛品种75头个体基因组重测序数据,用于分析涪陵水牛与西南地区其他水牛品种间的差异。其中8个品种68头沼泽型水牛样本信息为:滇东南水牛(DDN)10头、德宏水牛(DH)8头、盐津水牛(YJ)10头、宜宾水牛(YB)10头、贵州水牛(GZ)9头、贵州白水牛(GZB)12头、富钟水牛(FZ)6头、涪陵水牛3头(FL_35,FL_42,FL_131);3个品种7头河流型水牛样本为:摩拉水牛(ML)2头、尼里-拉菲水牛(NNLF)1头、槟榔江水牛(BLJ)4头。
1.2 试验方法
1.2.1 测序数据质控及比对参考基因组
本试验采集的27头水牛耳组织样本送往北京诺禾致源科技股份有限公司基于Illumina HiSeq PE150平台进行二代全基因组重测序。对本试验新测序原始数据Raw data和NCBI下载数据进行质控:1)去除带接头的reads pair;2)去除N的含量超过read长度比例的10%;3)去除低质量(Q≤5)碱基数超过read长度比例的 50%,得到高质量的Clean data。利用BWA(V0.7.17)软件(参数:mem -t 4 -k 32 -M)将Clean data比对到河流型水牛参考基因组(UOA_WB_1,GCF_003121395.1)上。
1.2.2 SNP检测及质控
利用SAMTOOLS(V1.9)软件检测群体SNP,按以下过滤条件进行质量控制:dp3(保留深度不低于3×的数据)、Miss0.2(保留缺失率不高于0.2的数据)、maf0.05(保留最小等位基因频率不小于0.05的数据),保留的位点用于后续分析。
1.2.3 涪陵水牛与西南地区水牛品种的遗传背景分析
通过 GCTA(V1.26)[18]软件进行主成分分析(PCA)并利用R(V4.2.2)软件绘图。祖先成分分析时,首先创建Plink(V1.9)的输入文件-Ped文件,然后利用ADMIXTURE[19]软件构建群体遗传结构,K取值为2~10。进化树分析采用Neighbor-joining法构建,利用 MEGA(V7.0)软件进行绘图。
1.2.4 保种场内涪陵水牛与毗邻区域水牛品种连续纯合片段(ROH)分析
基于滑动窗口的方法检测全基因组中的ROH。通过应用以下参数和阈值来定义ROH:--homozyg-density 500,--homozyg-window-het 1,--homozyg-window-snp 50,--homozyg-kb 1000,--homozyg-snp 30,统计全基因组ROH数目、长度、分布,计算ROH总长度与常染色体总长度的比值得到近交系数FROH,公式如下:
FROH=∑LROHLauto
其中,∑LROH为ROH片段的长度之和,Lauto为常染色体的总长度。
2 结 果
2.1 涪陵水牛基因组重测序及SNPs鉴定
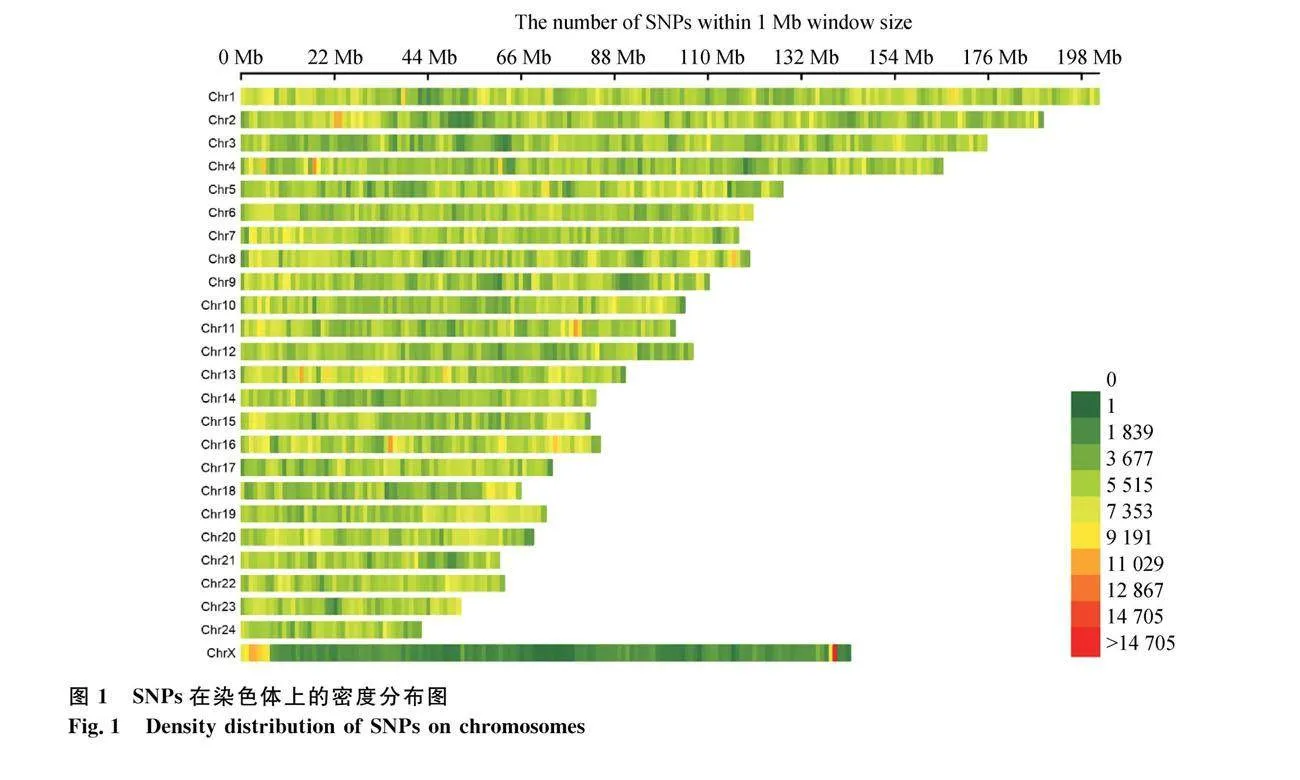
本研究新测序共产生766.402 G Raw data,过滤后的Clean data共764.202 G,GC含量在43.3%~44.05%之间。与参考基因组的平均比对率为99.15%,平均覆盖深度为10×,1×覆盖度在98.43%以上,测序质量高(Q30≥92.33%)。基于高质量河流型水牛(2n=50)参考基因组对涪陵水牛进行全基因组水平的SNP鉴定。通过质量控制后共鉴定到14 326 437个SNPs,未定位到染色体上的SNPs数为20 069个。所鉴定的SNPs在染色体上分布广泛(图1),1号染色体最长,SNPs位点最多,为1 176 548个,密度为5.82个·kb-1;24号染色体的SNP数量最少,为235 099个,密度为5.54个·kb-1;16号染色体所含有的SNP密度最高,为6.21个·kb-1。
2.2 涪陵水牛与西南地区水牛品种遗传背景分析
2.2.1 主成分分析
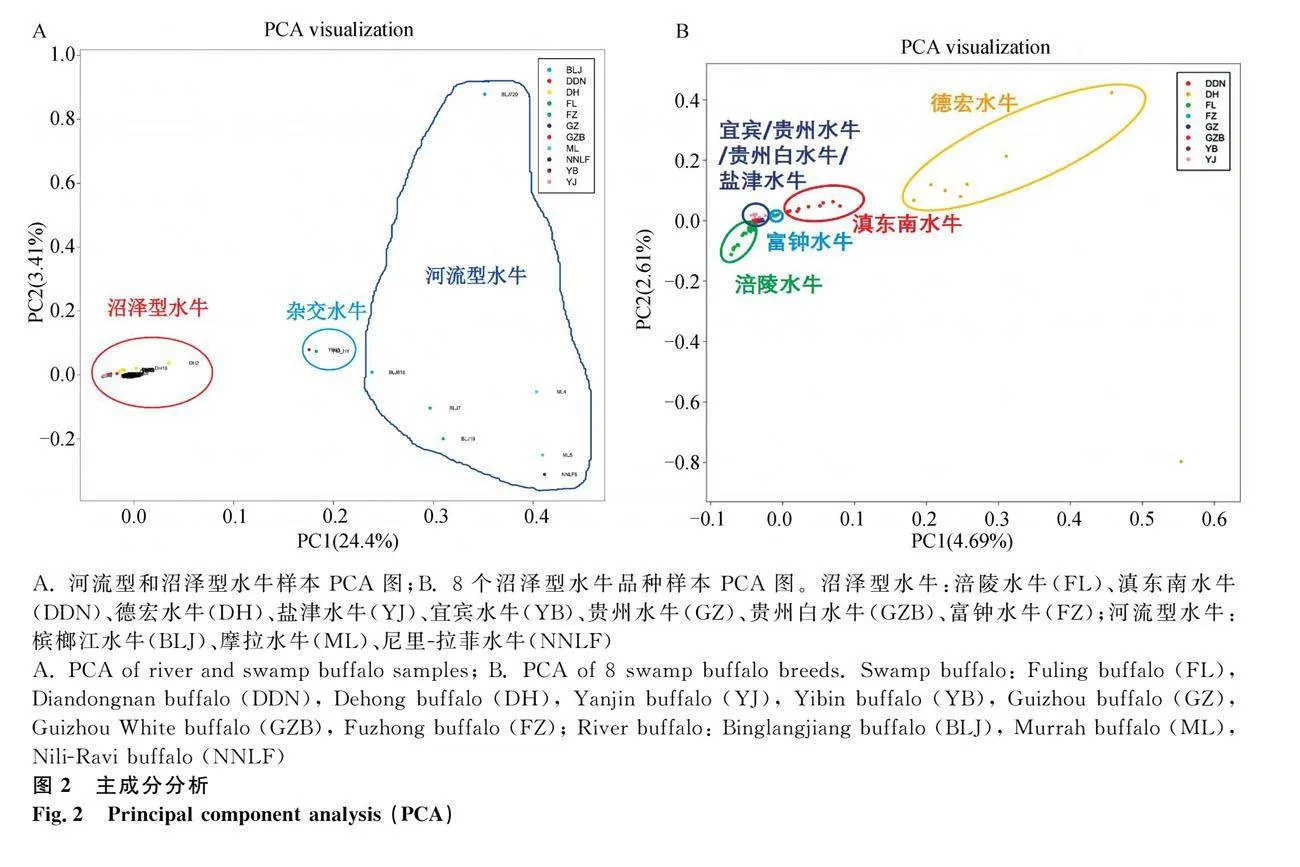
为检验样本的遗传背景,首先将27头水牛重测序个体以及NCBI下载的河流型水牛和沼泽型水牛样本进行PCA分析。如图2A所示,河流型水牛品种(BLJ、ML和NNLF)和沼泽型水牛(FL、DH、DDN、YB、GZ、GZB、YJ和FZ)间按照第一主成分(24.4%)区分明显;并且摩拉水牛与涪陵水牛的杂合个体(FM_HY)和一个NCBI下载的宜宾水牛(YB21)位于两类型水牛之间,在后续分析中将剔除这两个杂合个体。
进一步将沼泽型水牛个体进行PCA分析发现(图2B),德宏水牛(DH)、滇东南水牛(DDN)、涪陵水牛(FL)、富钟水牛(FZ)间按照第一主成分(4.69%)和第二主成分(2.61%)可以较好的区分;而宜宾水牛、贵州水牛、贵州白水牛、盐津水牛比较聚集。因此,涪陵水牛与毗邻区域的宜宾水牛、贵州水牛、贵州白水牛、盐津水牛间具有明显的区分,并且涪陵水牛个体较分散可分成2群。
2.2.2 群体遗传结构分析
系统发育树(Neighbor-joining法)结果显示(图3A),滇东南水牛、德宏水牛、盐津水牛、贵州白水牛、涪陵水牛有较好的区分度,涪陵水牛可明显分为两群;部分贵州水牛与贵州白水牛、宜宾水牛与NCBI下载的2头涪陵水牛(FL_35和FL_131)间存在混群现象。同时根据NJ树可以看出,FL22-26这几头犊牛与其母亲各自聚在一起,可对系谱记录进行纠偏。
群体祖先成分分析发现,最佳分群数为K=6(图3B),此时德宏水牛和盐津水牛各具有独立的一支结构,贵州白水牛大部分个体也具有独特群体结构,滇东南水牛与富钟水牛相似,贵州水牛、宜宾水牛和部分涪陵水牛具有一类相似结构,但可以明显看出部分涪陵水牛个体具有独特的一类遗传结构。
2.3 保种场内涪陵水牛与毗邻区域水牛品种群体ROH比较分析
2.3.1 ROH检测
由于涪陵水牛与毗邻区域地区地方水牛品种之间存在遗传差异,对涪陵水牛、宜宾水牛和贵州水牛进行ROH检测,比较涪陵水牛与其它两个品种的ROH数量以及长度的差异(图4A和4B)。在3个品种全基因组中共检测到2 722个ROHs,其中,在21头涪陵水牛中检测到1 593个ROHs,宜宾水牛(n=9)有592个ROHs,贵州水牛(n=9)有537个ROHs。
图4A展示了不同水牛群体的ROH在不同分类长度中的比例,贵州水牛的短ROH(1~2 Mb)片段较多,占贵州水牛所有ROH总数的86.78%,涪陵水牛短ROH片段占总个数的78.15%。涪陵水牛群体检测到8个长ROH(gt;16 Mb)片段,最长片段为24.56 Mb。而其他两组群体均没有长ROH片段检出。
在涪陵水牛中,检测到每头水牛平均75.85±12.53个ROHs,范围在50~100个,其余两个牛群中检测到每头水牛平均69.79±13.92个ROHs,范围在37~86个。3个牛群每头水牛所有ROH长度的平均值为(126.03±40.57) Mb,最小值和最大值分别为53.83和216.01 Mb。从图4B中可以明显看出,涪陵水牛个体的ROH总数和总长度较其他两组群体高。总的来看,ROH总长度有50~150 Mb的个体较多,而涪陵水牛个体ROH总长度主要分布在100~200 Mb之间。
2.3.2 基于ROH的群体近交程度分析
为了解群体的近交水平,对保种群涪陵水牛与毗邻区域宜宾水牛和贵州水牛群进行了基于全基因组ROH的近交系数计算,从图4C中可以明显看出涪陵水牛的FROH总体水平高于其他两组水牛群。保种场的涪陵水牛平均近交系数最高,为0.056 8,个体间差异较小,范围在0.036 5~0.082 4。宜宾水牛平均近交系数为0.041 0,范围在0.025 2~0.059 4,贵州水牛平均近交系数为0.034 6,范围在0.020 5~0.048 9。
半同胞交配产生后代的近交系数为0.125,涪陵水牛所有个体近交系数均低于0.125(表1),近交系数在0.062 5~0.125之间的个体为FL3、FL4、FL7、FL8、FL13、FL16、FL17,这7头涪陵水牛近交系数较高,存在近交风险。
3 讨 论
基于全基因组SNPs信息开展群体遗传多样性、群体结构和亲缘关系等分析,已被广泛应用于猪[20-22]、鸡[23-25]、鸭[26]、牛[27]、羊[28-30]等多个畜禽动物中。本研究同样基于SNP数据对26头涪陵水牛以及下载的68头西南地区沼泽型水牛和7头河流型水牛进行了群体遗传结构的分析。不同的是,特意引入河流型(摩拉水牛)和沼泽型(涪陵水牛)的杂合个体(FM_HY)用于检验样本的遗传背景。PCA分析显示沼泽型水牛和河流型水牛两个亚群明显分隔开(图2A)。但来源于Sun等[31]所测序的宜宾水牛YB21个体与已知杂合个体FM_HY距离近,且位于沼泽型与河流型水牛之间,这可能可以解释该论文中为什么部分南方水牛个体含有河流型水牛祖先成分。这提示,在进行群体遗传结构分析时,有必要先进行个体遗传背景的筛选。
河流型和沼泽型水牛间的群体遗传结构差异得到广泛关注[31-32]。Sun等[31]将德宏水牛和滇东南水牛划分为中国西南类群,将涪陵水牛、宜宾水牛、贵州水牛、贵州白水牛划为长江上游类群;而Luo等[32]将槟榔江水牛、德宏水牛划为河流型水牛与沼泽型水牛的杂交区,认为德宏水牛含有河流型水牛的遗传成分。本研究第一个核心目标是探索涪陵水牛是否与毗邻区域水牛具有遗传结构差异。为此,对8个西南地区沼泽型水牛进行PCA、系统发育树、祖先成分分析,结果显示德宏水牛和滇东南水牛相比西南地区其他6个水牛品种的确具有明显的群体结构差异。对分布于长江上游的涪陵水牛、宜宾水牛、贵州水牛、贵州白水牛进行更为细致的分析显示,涪陵水牛可分为2个类群,相比其他毗邻区域品种具有一类独特的遗传结构。后续有必要对涪陵水牛基因组结构进行深入研究。
长ROH和短ROH可以分别反映较近时期的近交活动(5代以前)和较远时期的近交活动(最多50代以前)[33-34]。Liu等[35]使用中密度SNP芯片分析了899头意大利地中海水牛全基因组ROH模式和近交水平,共鉴定出42 433个ROHs片段,以1~4 Mb为主的短片段约占全部短片段的72.29%。相比之下,较大的ROH片段(gt;8 Mb)仅占所有ROH片段的7.97%,基于ROH的近交系数(FROH)估计在0.020 1~0.037 1之间,表明意大利地中海水牛种群受到历史上近交事件的影响,但种群近期的近交水平较低。基于ROH计算基因组近交系数的方法能为遗传育种提供新的见解,且更能反映群体真实的近交水平[36-38]。Ghoreishifar等[39]基于伊朗Azeri(n=252)和Khuzestani(n=113)河流型水牛SNP基因分型数据,使用FGRM、FHOM、FUNI以及FROH等不同方法估计近亲繁殖系数并比较4种方法的可靠性,发现使用基因组近交系数FROH估计近亲繁殖的结果最为可靠。本研究发现,在保种场涪陵水牛群体中,每头水牛平均含有75.85±12.53个ROHs,远高于上述国外水牛品种中的平均每头ROH数量;并且在涪陵水牛群体中检测到8个长ROH(gt;16 Mb)片段,最长为24.56 Mb,提示涪陵水牛近期的近交活动较多且可能存在共同祖先。涪陵水牛保种群平均近交系数为0.056 8,远低于半同胞交配产生的后代的近交系数,但7头涪陵水牛近交系数较高在0.062 5~0.125之间,存在近交风险。
目前,涪陵水牛存在品种数量少、保种难度大等问题,有必要采取有效的保种措施,并应加快涪陵水牛的基础研究工作。第一,涪陵水牛具有两个亚群,应尽快将保护区内牛只进行遗传结构鉴定,保种场内和保护区内按亚群进行有计划选种选配,防止群体混杂。第二,保种群内的7头涪陵水牛存在近交风险,可以通过引进新的血统,建立健全系谱记录和基因库管理体系,避免近交或过度集中某些优良基因。第三,基于SNP数据进行选择信号分析有助于挖掘与动物经济性状相关的基因,例如免疫、肉质、繁殖和热应激相关的候选基因[32,40-41]。与SNP相比,拷贝数变异(copy number variation, CNV)可能对畜禽的表型产生更大的影响[42],研究发现了可能与水牛的适应性、产奶、免疫力等相关的基因CNV[43-45]。因此,后续可以利用涪陵水牛已有的SNP数据进行选择信号分析挖掘出重要性状的候选基因,也可以进一步对涪陵水牛开展基因组CNV的研究,更深入的解析其基因组特征,从而筛选涪陵水牛中的优良个体,为涪陵水牛的保种和选育提供帮助。
4 结 论
本研究基于全基因组测序技术分析了保种场涪陵水牛及西南地区水牛品种间的遗传结构,结果表明保种场内涪陵水牛与毗邻区域水牛品种间具有明显的遗传结构差异;涪陵水牛可划分为2个类群,与毗邻区域水牛品种相比具有一类独特的群体结构。基于ROH的群体近交系数分析表明,7头涪陵水牛近交系数在0.062 5~0.125之间,存在近交风险。本研究可为今后涪陵水牛的资源保护与利用工作提供理论依据和基础数据。
参考文献(References):
[1] 国家畜禽遗传资源委员会.中国畜禽遗传资源志·牛志[M].北京:中国农业出版社,2011:335-338.
China National Commission of Animal Genetic Resources.Animal genetic resources in China,Bovines[M].Beijing:China Agriculture Press,2011:335-338.(in Chinese)
[2] 《重庆市畜禽遗传资源志》编写组.重庆市畜禽遗传资源志[M].重庆:重庆出版社,2013:65.
Chongqing Livestock and Poultry Genetic Resources Compilation Group.Animal genetic resources in Chongqing[M]. Chongqing: Chongqing Press,2013:65.(in Chinese)
[1] 国家畜禽遗传资源委员会办公室. 关于公布《国家畜禽遗传资源品种名录》的通知[J]. 中华人民共和国农业农村部公报, 2020(6): 36.
Office of Animal Genetic Resources Committee. Notice on National Breed List of Animal Genetic Resources[J]. Gazette of the Ministry of Agriculture and Rural Affairs of the People’s Republic of China, 2020(6): 36.(in Chinese)
[2] 《重庆市畜禽遗传资源志》编写组. 重庆市畜禽遗传资源志[M]. 重庆: 重庆出版社, 2013:65.
Compilation Group of Chongqing Animal Genetic Resources. Animal genetic resources in Chongqing[M]. Chongqing: Chongqing Press, 2013: 65.(in Chinese)
[3] 涪陵水牛育种概况[J].四川畜牧兽医,1976(2):42-46.
General situation of Fuling buffalo breeding[J].Sichuan Animal amp; Veterinary Sciences,1976(2):42-46.(in Chinese)
[4] 姚仕顺,王德绪.摩拉水牛与涪陵水牛杂交效果初报[J].四川畜牧兽医,1981(4):1-4.
YAO S S,WANG D X.Preliminary report on hybridization effect between Murrah buffalo and Fuling buffalo[J].Sichuan Animal amp; Veterinary Sciences,1981(4):1-4.(in Chinese)
[5] 田有庆,唐玉华,郭春华,等.涪陵水牛染色体组型分析[J].四川畜牧兽医,1984(4):6-8.
TIAN Y Q,TANG Y H,GUO C H,et al.Chromosome karyotype analysis of Fuling buffalo[J].Sichuan Animal amp; Veterinary Sciences, 1984(4):6-8.(in Chinese)
[6] 史荣仙,左福元,董学虎.四川水牛血液蛋白多态性研究[J].四川农业大学学报,1992(1):122-126.
SHI R X,ZUO F Y,DONG X H.Studies on blood protein polymorphism in Sichuan swamp buffaloes[J].Journal of Sichuan Agricultural University,1992(1):122-126.(in Chinese)
[7] 史荣仙,付茂忠,赖松家,等.长江流域水牛血液蛋白多态性研究[J].遗传,1995,17(1):7-11.
SHI R X,FU M Z,LAI S J,et al.Study on blood protein polymorphism of buffalo in Yangtze river basin[J].Hereditas (Beijing),1995,17(1):7-11.(in Chinese)
[8] 赖松家,史荣仙,郑维明.中国水牛血清淀粉酶多态性及型命名研究[J].四川农业大学学报,1995,13(2):203-207.
LAI S J,SHI R X,ZHENG W M.Study on serum amylase polymorphism and genetypes named of the buffaloes in China[J].Journal of Sichuan Agricultural University,1995,13(2):203-207.(in Chinese)
[9] 齐国强,昝林森,张桂香,等.中国部分地方水牛品种mtDNA D-loop区遗传多样性与起源研究[J].畜牧兽医学报,2008,39(1):7-11.
QI G Q,ZAN L S,ZHANG G X,et al.Mitochondrial DNA D-loop genetic diversity and origin of some Chinese domestic buffalo breeds[J].Acta Veterinaria et Zootechnica Sinica,2008,39(1):7-11.(in Chinese)
[10] 谢文美,苏 锐,李明晖,等.涪陵水牛mtDNA D-loop区遗传多样性研究[J].西北农业学报,2008,17(5):56-60.
XIE W M,SU R,LI M H,et al.Study on mtDNA D-loop genetic diversity in Fuling swamp buffalo[J].Acta Agriculturae Boreali-Occidentalis Sinica,2008,17(5):56-60.(in Chinese)
[11] LOPEZ B I M,AN N,SRIKANTH K,et al.Genomic prediction based on SNP functional annotation using imputed whole-genome sequence data in Korean Hanwoo cattle[J].Front Genet,2021,11:603822.
[12] DEMIR E,MORAVACˇGKOV N,KARSLI T,et al.Future perspective of NGS data for evaluation of population genetic structure in Turkish cattle[J].Acta Fytotechn Zootechn,2022,25(2):117-121.
[13] WU F,SUN H,LU S X,et al.Genetic diversity and selection signatures within Diannan small-ear pigs revealed by next-generation sequencing[J].Front Genet,2020,11:733.
[14] CEBALLOS F C,JOSHI P K,CLARK D W,et al.Runs of homozygosity:windows into population history and trait architecture[J].Nat Rev Genet,2018,19(4):220-234.
[15] ONZIMA R B,UPADHYAY M R,DOEKES H P,et al.Genome-wide characterization of selection signatures and runs of homozygosity in Ugandan goat breeds[J].Front Genet,2018,9:318.
[16] SHI L Y,WANG L G,LIU J X,et al.Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a Large White pig population[J].J Anim Sci Biotechnol,2020,11(1):46.
[17] MARRAS G,GASPA G,SORBOLINI S,et al.Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy[J].Anim Genet,2015,46(2):110-121.
[18] YANG J,LEE S H,GODDARD M E,et al.GCTA:a tool for genome-wide complex trait analysis[J].Am J Hum Genet,2011, 88(1):76-82.
[19] LIU Y S,NYUNOYA T,LENG S G,et al.Softwares and methods for estimating genetic ancestry in human populations[J].Hum Genomics,2013,7(1):1.ALEXANDER D H, LANGE K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation[J]. BMC Bioinformatics, 2011, 12: 246.
[20] 赵真坚,王书杰,陈 栋,等.基于低深度全基因组测序分析内江猪群体结构和遗传多样性[J].畜牧兽医学报,2023,54(6):2297-2307.
ZHAO Z J,WANG S J,CHEN D,et al.Population structure and genetic diversity analysis of Neijiang pigs based on low-coverage whole genome sequencing[J].Acta Veterinaria et Zootechnica Sinica,2023,54(6):2297-2307.(in Chinese)
[21] 陶 璇,杨雪梅,梁 艳,等.基于SNP芯片的丫杈猪保种群体遗传结构研究[J].畜牧兽医学报,2023,54(6):2308-2319.
TAO X,YANG X M,LIANG Y,et al.Analysis of genetic structure of conservation population in Yacha pig based on SNP chip[J].Acta Veterinaria et Zootechnica Sinica,2023,54(6):2308-2319.(in Chinese)
[22] 胡紫平,王立刚,宗文成,等.基于基因组SNP和ROH的剑白香猪群体遗传结构解析[J].畜牧兽医学报,2023,54(10):4117-4125.
HU Z P,WANG L G,ZONG W C,et al.Genetic structure analysis of Jianbai Xiang pig population based on genomic SNP and ROH[J].Acta Veterinaria et Zootechnica Sinica,2023,54(10):4117-4125.(in Chinese)
[23] 齐丽娜,陆雪林,杨凯旋,等.基于SNP芯片分析新浦东鸡的遗传多样性和遗传结构[J].畜牧兽医学报,2023,54(12):4962-4971.
QI L N,LU X L,YANG K X,et al.Analysis of genetic diversity and genetic structure of new Pudong chicken based on SNP chips[J].Acta Veterinaria et Zootechnica Sinica,2023,54(12):4962-4971.(in Chinese)
[24] 武艳平,魏 岳,康昭风,等.基于全基因组SNP分析8个地方鸡品种的遗传多样性[J].畜牧兽医学报,2022,53(2):646-653.
WU Y P,WEI Y,KANG Z F,et al.Genetic diversity analysis of 8 local chicken breeds based on whole genome SNP[J].Acta Veterinaria et Zootechnica Sinica,2022,53(2):646-653.(in Chinese)
[25] 高超群,曹然然,杜文苹,等.基于全基因组SNP标记分析中国地方鸡品种的遗传多样性和种群结构[J].畜牧兽医学报,2023,54(2):554-562.
GAO C Q,CAO R R,DU W P,et al.Genetic diversity and population structure analysis of Chinese native chicken breeds using genome-wide SNPs[J].Acta Veterinaria et Zootechnica Sinica,2023,54(2):554-562.(in Chinese)
[26] 刘宏祥,沈永杰,张丽华,等.基于简化基因组测序的娄门鸭遗传多样性评价[J].畜牧兽医学报,2022,53(6):1735-1748.
LIU H X,SHEN Y J,ZHANG L H,et al.Genetic diversity evaluation of Loumen duck based on reduced-representation genome sequencing[J].Acta Veterinaria et Zootechnica Sinica,2022,53(6):1735-1748.(in Chinese)
[27] 马浩然,张路培,金生云,等.利用高密度SNP芯片评估中国地方肉牛品种基因组亲缘关系[J].畜牧兽医学报, 2023,54(10):4174-4185.
MA H R,ZHANG L P,JIN S Y,et al.Assessment of the genomic relationships for Chinese indigenous beef cattle using high-density SNP chip[J].Acta Veterinaria et Zootechnica Sinica,2023,54(10):4174-4185.(in Chinese)
[28] 马克岩,韩金涛,白雅琴,等.基于简化基因组测序的永登七山羊遗传多样性分析[J].畜牧兽医学报,2023,54(5):1939-1950.
MA K Y,HAN J T,BAI Y Q,et al.Genetic diversity analysis of Yongdeng Qishan sheep based on specific-locus amplified fragment sequencing[J].Acta Veterinaria et Zootechnica Sinica,2023,54(5):1939-1950.(in Chinese)
[29] 张任豹,周东辉,周李生,等.基于70 K SNP芯片分析济宁青山羊保种群体的遗传结构[J].畜牧兽医学报,2023,54(7):2836-2847.
ZHANG R B,ZHOU D H,ZHOU L S,et al.Analysis of genetic structure of conservation population in Jining gray goats based on 70 K SNP chip[J].Acta Veterinaria et Zootechnica Sinica,2023,54(7):2836-2847.(in Chinese)
[30] 李隐侠,牙生江·那斯尔,赛里克·都曼,等.SNP芯片评估柯尔克孜羊群体遗传多样性和遗传结构[J].畜牧兽医学报, 2023,54(2):572-583.
LI Y X,NASIER Y,DUMAN S,et al.Evaluation of genetic diversity and genetic structure in Kirgiz sheep population based on SNPs chip[J].Acta Veterinaria et Zootechnica Sinica,2023,54(2):572-583.(in Chinese)
[31] SUN T,SHEN J F,ACHILLI A,et al.Genomic analyses reveal distinct genetic architectures and selective pressures in buffaloes[J]. Gigascience,2020,9(2):giz166.
[32] LUO X E,ZHOU Y,ZHANG B,et al.Understanding divergent domestication traits from the whole-genome sequencing of swamp- and river-buffalo populations[J].Natl Sci Rev,2020,7(3):686-701.
[33] FERENACˇGAKOVIC' M,SLKNER J,CURIK I.Estimating autozygosity from high-throughput information:effects of SNP density and genotyping errors[J].Genet Sel Evol,2013,45(1):42.
[34] MASTRANGELO S,CIANI E,MARSAN P A,et al.Conservation status and historical relatedness of Italian cattle breeds[J].Genet Sel Evol,2018,50(1):35.
[35] LIU S H,MA X Y,HASSAN F U,et al.Genome-wide analysis of runs of homozygosity in Italian Mediterranean buffalo[J].J Dairy Sci,2022,105(5):4324-4334.
[36] PURFIELD D C,BERRY D P,MCPARLAND S,et al.Runs of homozygosity and population history in cattle[J].BMC Genet,2012, 13(1):70.
[37] PERIPOLLI E,MUNARI D P,SILVA M V G B,et al.Runs of homozygosity:current knowledge and applications in livestock[J]. Anim Genet,2017,48(3):255-271.
[38] PERIPOLLI E,STAFUZZA N B,MUNARI D P,et al.Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle[J].BMC Genomics,2018,19(1):34.
[39] GHOREISHIFAR S M,MORADI-SHAHRBABAK H,FALLAHI M H,et al.Genomic measures of inbreeding coefficients and genome-wide scan for runs of homozygosity islands in Iranian river buffalo,Bubalus bubalis[J].BMC Genet,2020,21(1):16.
[40] RAFIEPOUR M,EBRAHIMIE E,VAHIDI M F,et al.Whole-genome resequencing reveals adaptation prior to the divergence of buffalo subspecies[J].Genome Biol Evol,2021,13(1):evaa231.
[41] NASCIMENTO A V,CARDOSO D F,SANTOS D J A,et al.Inbreeding coefficients and runs of homozygosity islands in Brazilian water buffalo[J].J Dairy Sci,2021,104(2):1917-1927.
[42] MILLS R E,WALTER K,STEWART C,et al.Mapping copy number variation by population-scale genome sequencing[J].Nature, 2011,470(7332):59-65.
[43] STRILLACCI M G,MORADI-SHAHRBABAK H,DAVOUDI P,et al.A genome-wide scan of copy number variants in three Iranian indigenous river buffaloes[J].BMC Genomics,2021,22(1):305.
[44] ZHANG X F,CHEN N B,CHEN H,et al.Comparative analyses of copy number variations between swamp and river buffalo[J].Gene,2022,830:146509.
[45] YANG L,HAN J Z,DENG T X,et al.Comparative analyses of copy number variations between swamp buffaloes and river buffaloes[J].Anim Genet,2023,54(2):199-206.
(编辑 郭云雁)
