
利用GWAS和DNA甲基化共定位鉴定猪肉质性状的候选基因
2024-09-19 00:00:00崔晟頔王凯赵真坚陈栋申琦余杨王俊戈陈子旸禹世欣陈佳苗王翔枫,唐国庆
畜牧兽医学报 2024年5期





摘 要: 为了更深入地了解肉质性状的遗传基础,本研究使用基于基因型填充的测序数据进行了GWAS分析并与之前的发表的EWAS结果进行共定位分析。该研究发现了515个全基因组水平显著的SNPs位点和4 134个达建议显著阈值的SNPs位点,这些位点共涉及406个基因,其中包括262个蛋白质编码基因,最终确定了6个可能与肉质性状相关的重要候选基因。同时,与EWAS的结果进行比较,发现有12个显著的CpG位点与显著SNP位点存在重叠。通过这些研究可以为改善猪肉品质的育种工作提供更坚实的理论基础和数据支持。
关键词: 全基因组关联分析;SNPs;肉质性状
中图分类号:S828.2
文献标志码:A""" 文章编号:0366-6964(2024)05-1945-13
收稿日期:2023-12-08
基金项目:四川省科技厅项目(2020YFN0024;2021ZDZX0008;2021YFYZ0030);国家生猪技术创新中心先导科技项目(NCTIP-XD/B01)
作者简介:崔晟頔(2000-),男,河南新乡人,硕士,主要从事猪遗传育种研究,E-mail:313164192@qq.com
*通信作者:唐国庆,主要从事猪分子数量遗传研究,E-mail:tyq003@163.com
Identification of Candidate Genes for Pork Texture Traits Using GWAS Combined with
Co-localisation of DNA Methylation
CUI" Shengdi1,2,3, WANG" Kai4, ZHAO" Zhenjian1,2,3, CHEN" Dong1,2,3, SHEN" Qi1,2,3, YU" Yang1,2,3,
WANG" Junge1,2,3, CHEN" Ziyang1,2,3, YU" Shixin1,2,3, CHEN" Jiamiao1,2,3, WANG" Xiangfeng1,2,3,
TANG" Guoqing1,2,3*
(1.State Key Laboratory of Swine and Poultry Breeding Industry,College of Animal
Science and Technology, Sichuan Agricultural University, Chengdu 611130," China;
2.Key Laboratory of Livestock and Poultry Multi-omics of Ministry of Agriculture and
Rural Affairs, College of Animal Science and Technology, Sichuan Agricultural University,
Chengdu 611130," China; 3.Farm Animal Genetic Resources Exploration and Innovation
Key Laboratory of Sichuan Province, Sichuan Agricultural University, Chengdu 611130,
China;
4.New Hope Liuhe CO. LTD., Chengdu 625014," China)
Abstract: To gain a deeper understanding of the genetic basis of meat quality traits, this study performed GWAS analysis using genotype-based populated sequencing data and co-localisation analysis with previously published EWAS results. The study identified 515 genome-wide significant SNP loci and 4 134 SNP loci at the proposed significance threshold, which involved 406 genes, including 262 protein-coding genes, and resulted in the identification of 6 important candidate genes that may be associated with meat quality traits. Meanwhile, comparison with the results of EWAS revealed that 12 significant CpG loci overlapped with significant SNP loci. The study results can provide a more solid theoretical foundation and data support for breeding efforts to improve pork quality.
Key words: genome-wide association study; SNPs; meat traits
*Corresponding author:TANG Guoqing, E-mail:tyq003@163.com
猪肉是我国肉类食品中的重要组成部分,长期以来在国内肉类消费中占据主导位置。随着经济的繁荣和人民生活水平的提高,消费者对优质猪肉的需求日益增长。在当前的研究中,随着高通量测序技术的广泛应用和低成本SNP芯片的开发,全基因组关联分析(GWAS)已成为研究家畜重要经济性状的有力工具[1]。通过进行GWAS分析,可以识别与肉质性状相关的SNP位点,并挖掘重要的候选基因。这些发现为提高猪肉质量的育种工作提供了重要的理论基础。
现有的研究越来越多地表明,单核苷酸多态性(SNP)对基因组中大量DNA的甲基化具有显著影响。虽然全基因组表观遗传关联研究(EWAS)可以揭示DNA甲基化与复杂表型之间的关系,但尚不能明确其与表型变异的因果关系。有研究表明,表观遗传变异可能受到SNP[2-3]、转录因子结合[4]或环境因素[5]的影响。因此,EWAS识别的显著相关性可能与其他因素混淆。为了深入了解肉质性状的遗传基础,本研究基于基因型填充测序数据对大白猪群体进行GWAS分析。一方面,识别与肉质性状显著相关的SNP位点,并挖掘影响肉质性状的重要候选基因;另一方面,与之前的EWAS分析结果进行比较,从基因组水平上验证EWAS识别的关键区域和候选基因。
1 材料与方法
1.1 试验动物与表型测定
在本研究选取了浙江天蓬猪场140头纯种大白猪(包括85头公猪和55头母猪)。所有试验猪都在相同的设施条件和环境下的圈舍中饲养,并执行相同的饲养标准。在屠宰时,这些猪的平均体重为(111.71±12.92) kg,并在屠宰前禁食了24 h。在屠宰过程中,从每头猪的左半边胴体第3~4肋处背最长肌收集样品,并对其pH、肉色、滴水损失、肌内脂肪和脂肪酸组成等肉质性状进行测定。
1.2 组织样本采集及DNA提取和SNP分型
在对试验猪进行屠宰测定的同时采集肌肉样品于冻存管中,加入 75% 的乙醇,于-20℃ 条件下保存备用。耳组织 DNA 的提取参照 OMEGA 组织 DNA 提取试剂盒说明书所述步骤进行。基于 Illumina 平台(Illumina,美国)标准流程使用 Neogen GeneSeek Porcine50K 芯片对 140 头大白猪进行基因分型,芯片包含50 697个SNPs。
1.3 基因型填充
1.3.1 基因型填充参考模板
本研究采用了两种参考模板对芯片数据进行填充,并将两种参考模板填充的结果进行了合并。第一种参考模板的参考群体来自华南农业大学的张哲课题组[6](包括50头来自福建某猪场的杜洛克,以及来自华南地区21个地方猪品种的210头猪),芯片数据填充过程由对方完成。第二种参考模板的参考群体则是本课题组搜集的40头大白猪的重测序数据。
1.3.2 基因型填充及质量控制
填充完成后利用 Plink v1.09 软件[7]对填充后的 SNP 数据进行质量控制,过滤标准为:1)去除实际等位基因与最可能等位基因相关性平方(R2)小于0.9的SNPs位点;2)去除MAF小于0.01的SNPs位点;3)去除HWE检验小于 1×10-6的SNPs位点;4)去除性染色体上的 SNPs位点。经过质量控制检验的SNPs位点被用于后续 GWAS 分析。
1.4 全基因组关联分析
本研究使用 GEMMA v0.98.5 软件[8]中的线性混合模型对每个肉质性状进行 GWAS 分析。在 GWAS 分析前,先使用 Box-Cox 变换将表型数据校正至符合正态分布,然后以出生年、出生月、性别、批次作为固定因子对表型进行校正,得到的校正值被用于 GWAS 分析。GWAS 所使用的线性混合模型如下:
y=Xm+Wa+e
模型中,y表示表型值向量;m表示 SNP 效应;a表示剩余多基因效应,服从(a~MVN(0,Aσ2e))分布,A表示分子亲缘关系矩阵 (genomic relatedness matrix,GRM) ;e表示残差,服从(e~MVN(0,Iσ2e))分布,I表示单位向量;X和W分别表示 m和a的关联矩阵。GRM 根据如下公式计算:
G=1p∑pi=1(xi-1nx-i)(xi-1nx-i)T
式中,G表示个体间亲缘关系矩阵;n 表示个体数量;p表示 SNP 的数量;i表示第i个SNP;X表示n×p的表型矩阵; xi表示第i个SNP 的基因型;x-i表示第i个SNP 的平均值。
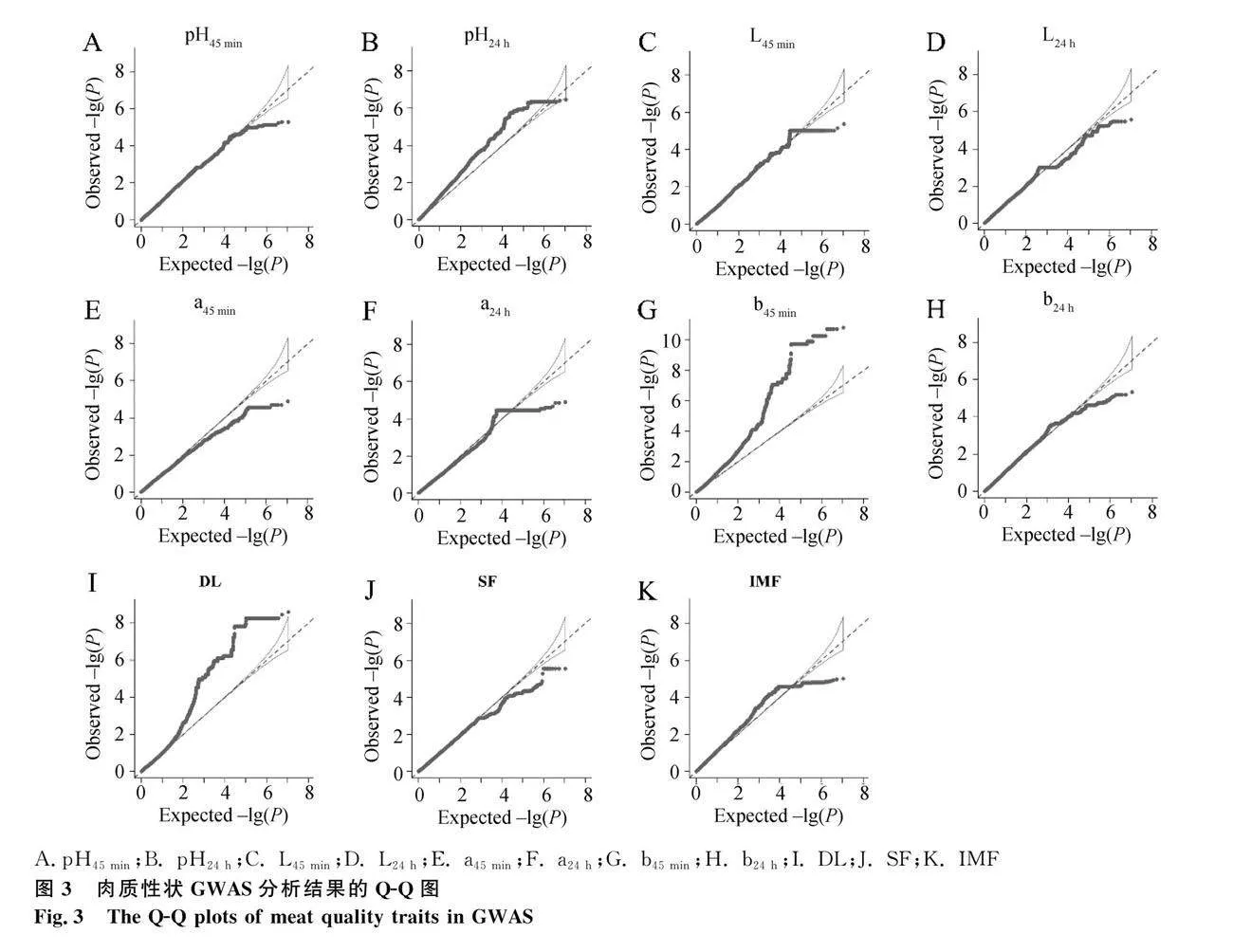
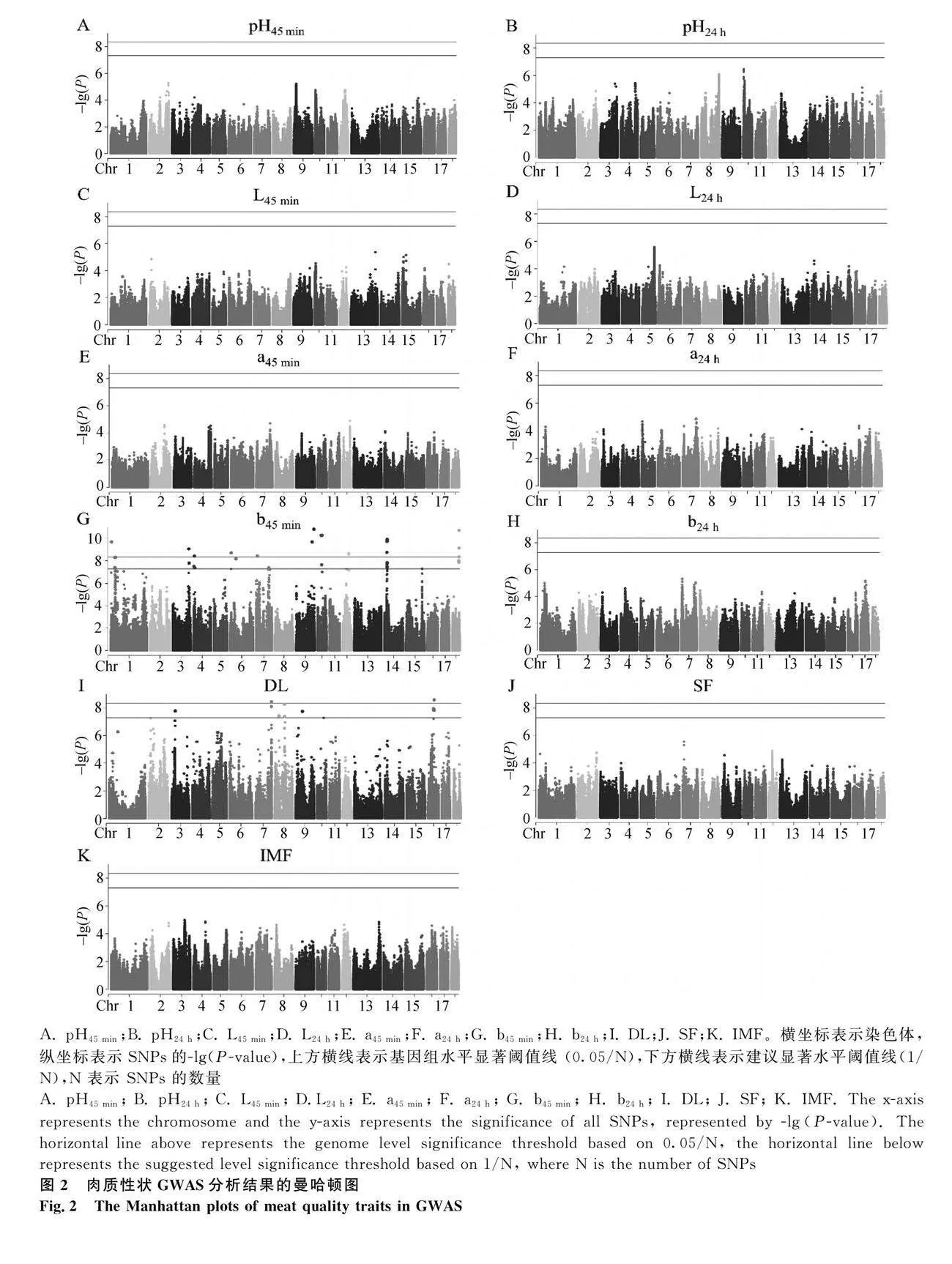
本研究使用Bonferroni校正法来设置全基因组显著阈值,即 GWAS 结果中当 SNP 的关联P值达到 Plt;0.05/N时,表示该 SNP 为显著 SNP 位点,其中 N表示 GWAS 分析中使用的 SNP 数量;建议显著阈值设置为Plt;1/N。然后使用R软件中的CMplot v4.0.0 包绘制曼哈顿图。使用R软件中GenABEL包[9]中的estlambda函数计算基因组膨胀系数 λ 用于判断是否有假阳性信号。
1.5 候选基因注释和功能分析
本研究中,以显著 SNP 位点上、下游 20 kb 范围的基因组区域作为 QTL 区域,根据 Ensembl 网站的 BioMart 模块检索范围内的候选基因,然后使用 DAVID 网站对候选基因进行 GO 和 KEGG 富集分析,并结合候选基因相关的已发表文献综合分析候选基因的生物学功能。
1.6 EWAS 与 GWAS 的共定位分析
本研究对 140 头大白猪群体肉质性状的 GWAS 鉴定的显著SNPs与EWAS(EWAS结果见文献[10])鉴定的显著CpG位点进行比较,从而鉴定GWAS与EWAS重叠的显著关联位点。如果GWAS的显著SNPs与EWAS显著CpG位点在2 Mb范围内,则被认为是二者重叠的显著关联位点[11]。
2 结 果
2.1 表型统计
本研究中 140 头大白猪(ZJYY)的表型统计见表1。
2.2 SNP数据填充统计结果
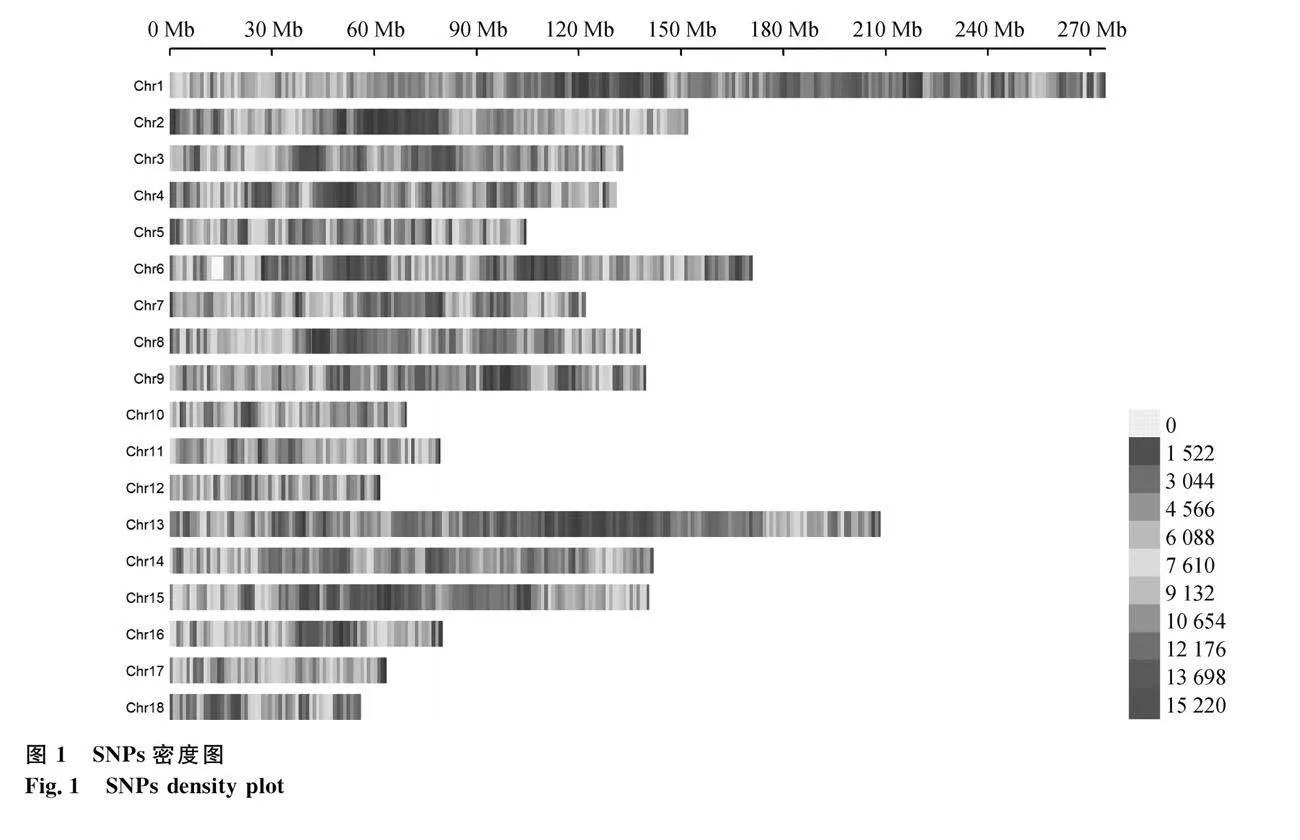
本研究对140头试验大白猪进行了50K芯片基因分型,并获得了50 697个SNPs。经过填充、合并及质量控制后,成功获得了10 961 097个高质量的SNPs(图1),这些SNPs被用于后续的GWAS研究。
2.2.1 肉质性状 GWAS 结果
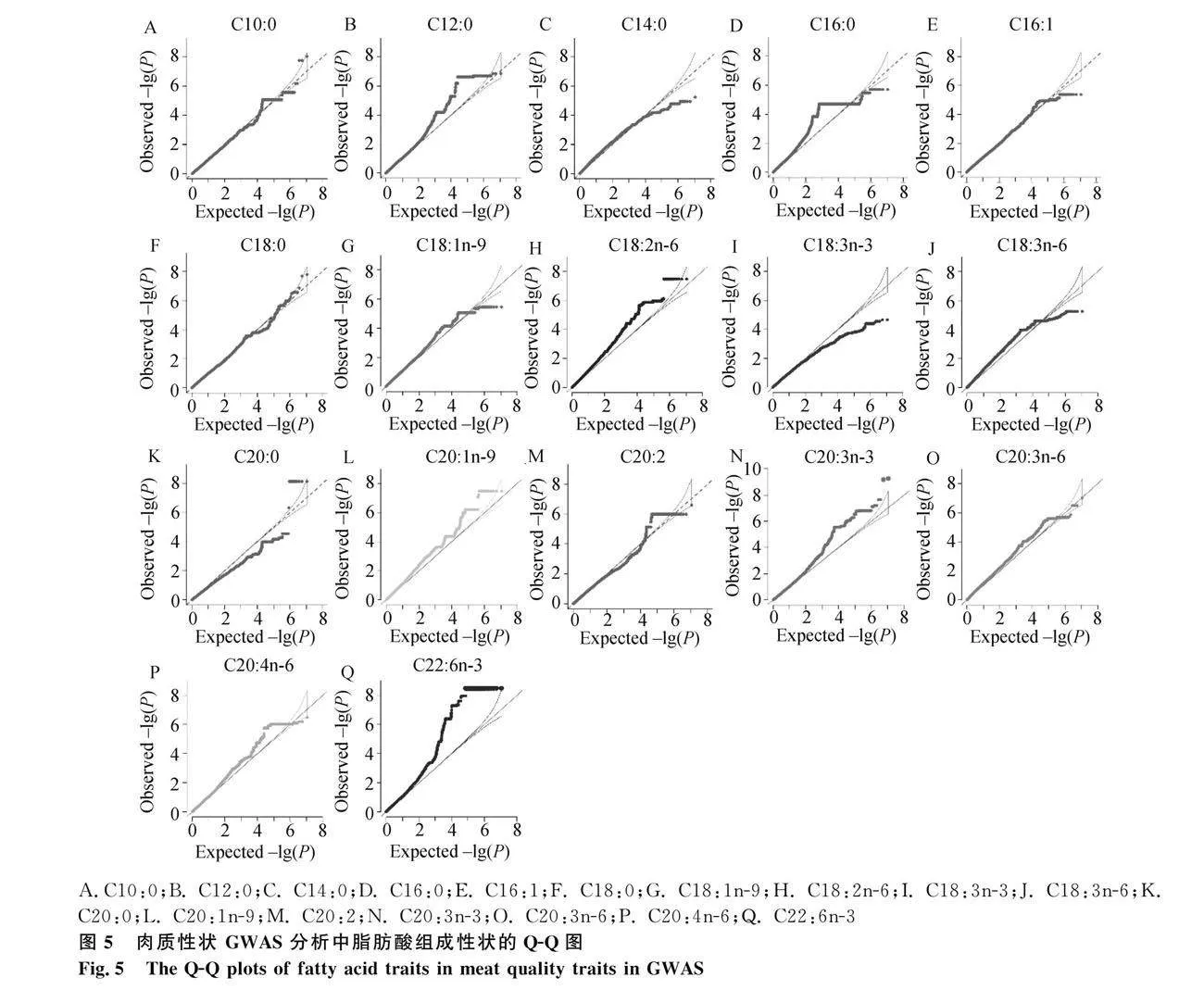
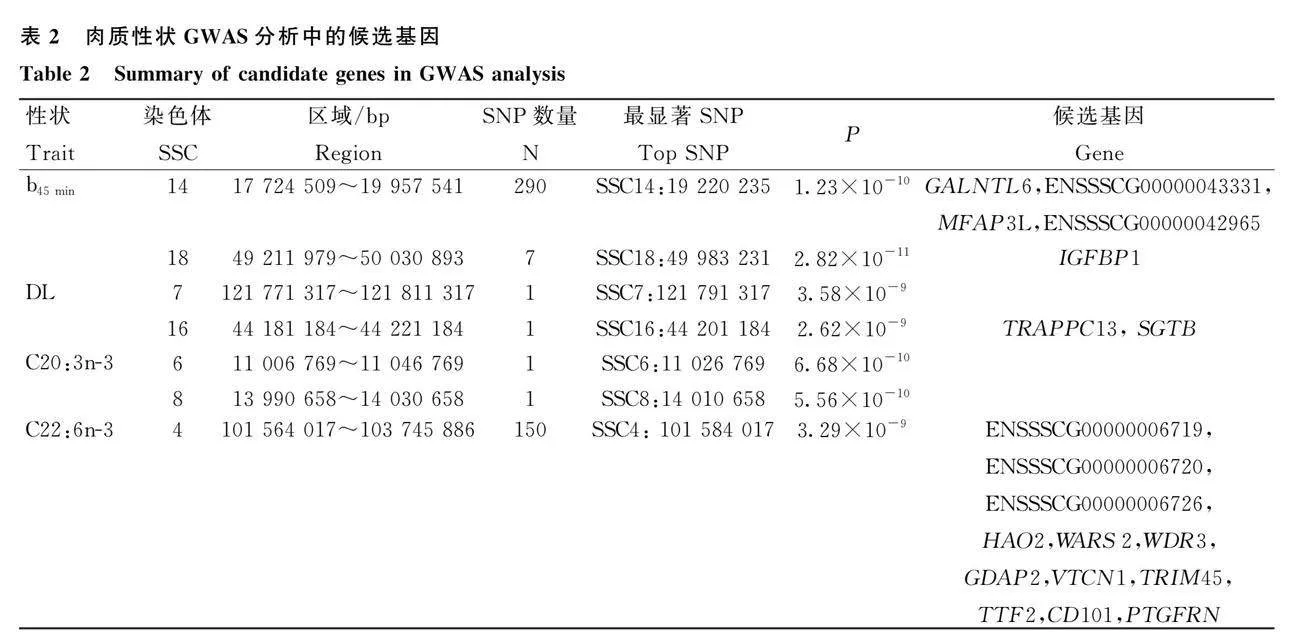
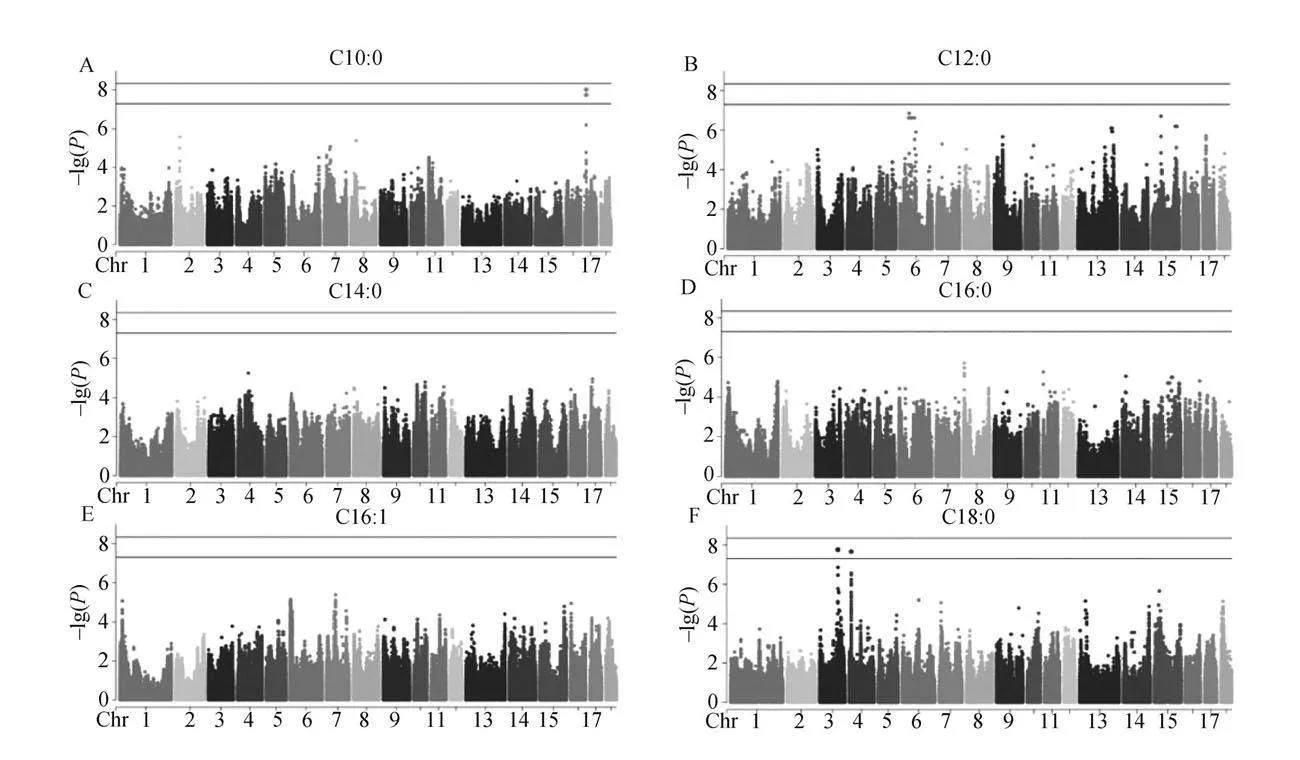
本研究对140头大白猪的28个肉质性状进行GWAS分析。结果显示,在所有肉质性状的分析中,有4个性状共检测到516个全基因组水平显著的SNPs(图2、图3、图4、图5)。其中,b45min性状的关联结果检测到最多的362个全基因组水平显著SNPs位点,其次是C22∶6n-3(150个)、DL(2个)以及C20∶3n-3(2个)。各性状间无共享SNPs,表明这几个肉质性状之间的SNP关联模式可能存在差异。本研究根据SNP位点的基因组位置,对516个全基因组水平显著的SNPs进行了注释,共注释了68个基因。其中包括34个蛋白质编码基因,详见表2。
此外,所有肉质性状 GWAS 结果中检测到共 4 134个达建议显著阈值的 SNPs 与 9个性状显著相关。其中 b45min性状结果检测到最多的 2 544个达建议显著阈值的 SNPs 位点,其次是 C22∶6n-3(1 069个)、DL(441个)、C20∶1n-9(28个)、C18∶2n-6(25个)、C20∶0(12个)、C20∶3n-3(10个)、C10∶0(3个)以及C18∶0(2个)。本研究基于 SNP 位点的基因组位置,对这4 134个达建议显著阈值的 SNPs进行了注释,共注释了338个基因,其中包括228个蛋白质编码基因。
2.3 候选基因注释及功能分析
为了进一步探究肉质性状候选基因的生物学功能,本研究对 GWAS 注释的 338个候选基因进行 GO 和 KEGG 分析。GO 分析结果表明:这些基因主要富集在金属离子结合、细胞间黏附、内质网功能、脂肪酸代谢、胰岛素代谢、氧化还原酶活性以及辅酶活性等相关GO条目中(图6A)。KEGG 富集分析结果表明:这些候选基因主要参与叶酸生物合成、脂肪酸延长、花生四烯酸代谢、泛酸和辅酶 A 的生物合成以及细胞色素对外源物代谢等通路(图6B)。这些结果表明,大部分候选基因可能通过调控离子结合、内质网功能以及脂肪酸代谢等相关生物学通路来影响肉质性状。
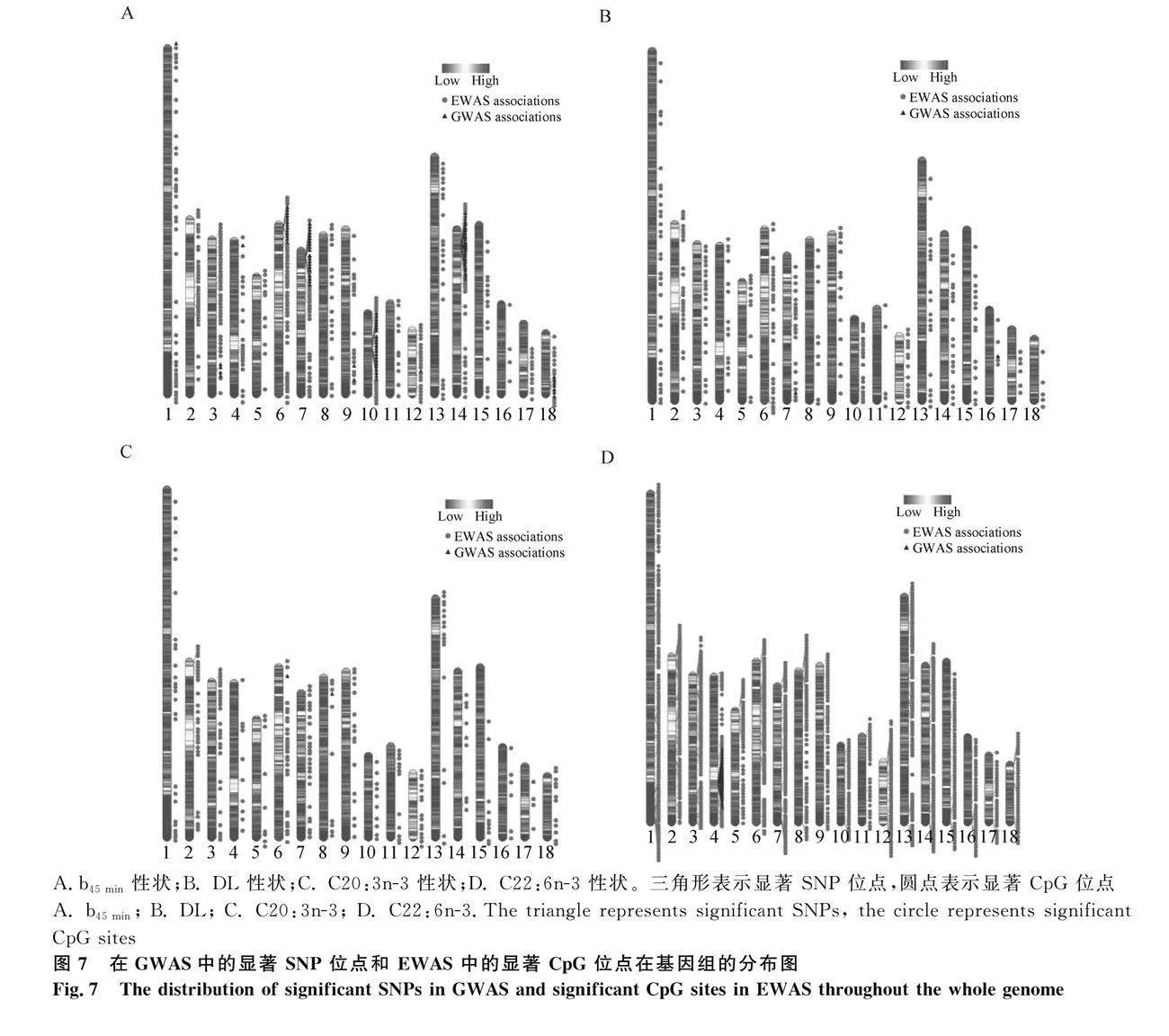
2.4 EWAS 与 GWAS 共定位分析
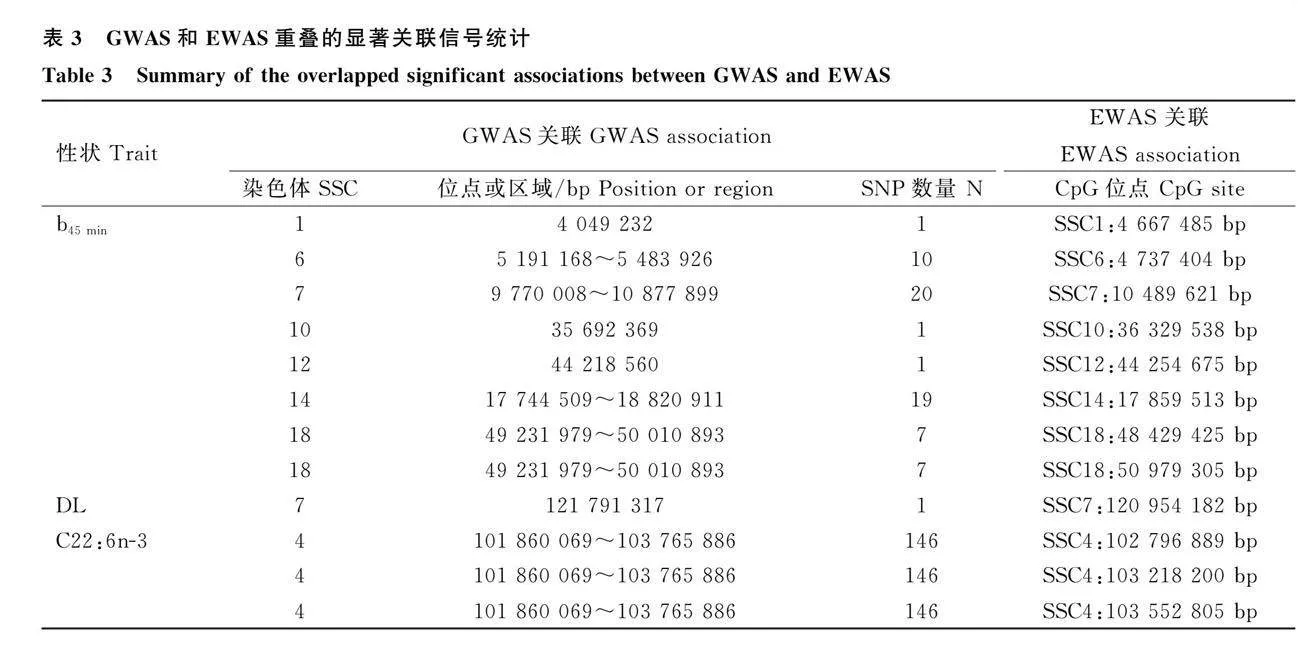
在本研究中,如果 GWAS 的显著 SNP 位点与 EWAS 显著 CpG 位点距离在 2Mb 范围内,则被认为是 GWAS 与 EWAS 重叠的显著关联位点。基于此标准,在研究的 28个肉质性状中,在 3个性状(b45min、DL 和 C22∶6n-3)上发现了GWAS与EWAS 重叠的显著关联位点。结果如图7和表3所示。其中,在 b45min中发现了 8个显著 CpG 位点和59个显著 SNPs 位点位置接近;在DL发现 1个CpG位点和1个SNP位点位置接近;在C22∶6n-3中发现了 3个CpG位点与 146个SNPs位点位置接近。这些位点被确定为重叠的显著关联位点。
3 讨 论
3.1 基于 GWAS 发现与肉质性状相关的 QTL
本研究基于GWAS对大白猪的28个肉质性状进行了关联分析,在全基因组水平上检测到516个显著的SNPs位点,并鉴定了3个与肉质性状相关的QTL区域。在b45min性状的关联结果中,SSC14:17.74~19.94 Mb区域包含290个显著的SNP。与猪QTL数据库的比较发现,该基因组区域附近的大多数QTL都与猪的生长性状有关。例如,Ma等[12]在白杜洛克×二花脸猪群体研究中报道了该区域附近与眼肌面积相关的QTL。此外,SSC18:49.28~50.01 Mb区域包含7个显著的SNPs。该基因组区域附近研究的大多数QTL都与胴体性状有关。例如,Liu等[13]在杜洛克×皮特兰群体的研究中鉴定了与DL性状相关的QTL。
在C22∶6n-3的关联结果中,SSC4∶101.55~103.75 Mb区域包含150个显著的SNPs。Prez-Enciso等[14]报道了SSC4上与脂肪酸代谢相关的QTL。Uemoto等[15]在纯种杜洛克群体的研究中也发现了与SSC4上的C18∶0显著相关的QTL。
3.2 重要的候选基因
本研究通过 GWAS对大白猪群体进行了肉质性状相关重要候选基因的鉴定与发掘,共鉴定了68个候选基因。通过功能富集分析以及查阅相关文献,确定了6个可能与肉质性状相关的重要候选基因,包括 NDUFB6、 GALNTL6、IGFBP1、ZFAT、CDH13 以及 MCUR1。
NDUFB6 编码线粒体复合物Ⅰ亚基,该基因在线粒体呼吸中发挥重要作用[16-17]。研究表明,线粒体呼吸和肌红蛋白的相互作用对畜禽屠宰前后的肉质均有影响[18]。此外,研究还发现线粒体能调控低氧条件下肌红蛋白的表达,而且线粒体及其中间产物对肌肉中肌红蛋白的还原具有积极作用[19]。因此,NDUFB6基因可能通过影响线粒体功能来调节肌红蛋白的表达,进而对肉质性状产生影响。
GALNTL6主要编码多肽 N-乙酰半乳糖胺基转移酶样6,并且在一项针对荷斯坦牛的 GWAS 分析中,发现该基因与牛胴体性状存在关联[20]。
IGFBP1属于胰岛素样生长因子家族,有研究表明,胰岛素样生长因子系统在猪骨骼肌生长中起着重要作用[21]。此外,胰岛素样生长因子信号通路在调节骨骼肌生长、分化和维持成人肌肉组织的稳态方面起着关键作用[22]。因此,IGFBP1可能通过参与骨骼肌生长分化而对肉质性状产生影响。
ZFAT编码一种锌指蛋白,Ishikura等[23]的研究发现 ZFAT 在脂肪细胞的维持和分化中具有至关重要的作用。脂肪沉积与肉品质密切相关,研究表明肌肉的IMF可以影响肉的风味、嫩度以及多汁性,而且脂肪沉积量也直接影响肉色、滴水损失以及大理石花纹[24]。
CDH13编码钙粘蛋白超家族的成员,即钙依赖性细胞粘附蛋白。基于大白猪基因型填充数据的一项一步法GWAS研究表明,CDH13与猪的体型性状存在关联[25]。
MCUR1在线粒体能量代谢中具有重要调节作用[26]。研究表明,MCUR1是线粒体 Ca2+摄取和维持正常细胞生物能所需的线粒体单向转运通道复合体的关键组成部分[27]。鉴于线粒体功能对肉质的重要影响,MCUR1基因可能通过参与线粒体功能而对肉质性状产生影响。
3.3 肉质性状的 GWAS 与 EWAS 共定位比较
通过比较 EWAS 鉴定的显著 DNA 甲基化位点与 GWAS 鉴定的显著 SNPs 之间的关联模式,发现在 EWAS 分析中鉴定的显著关联在整个基因组更加分散,大多数显著关联局限于单个 CpG 位点,此外,许多 CpG 位点的 DNA 甲基化与多个肉质性状相关,而诸如 b45min、DL 和 C22∶6n-3 等性状与位于不同染色体的多个CpG位点相关。相比之下,由于基因组上存在大量的连锁不平衡(LDs)以及SNP在基因组上密度更高,GWAS分析中鉴定的显著关联往往延伸几kb甚至几Mb。DNA甲基化的关联模式反映了不同染色体区域中DNA甲基化的调控特性,这是不同基因组区域中DNA甲基化对基因表达的调控不同所致。
在GWAS对肉质性状的分析中,鉴定的显著关联主要涉及 b45min、DL、C20∶3n-3和C22∶6n-3 等性状。在EWAS分析中,也鉴定了这些性状的大量显著关联信号。此外,这些性状的显著SNP位点和显著CpG位点在基因组中的位置也有一些比较接近。许多研究表明,遗传变异可以对DNA甲基化组的大部分产生重要影响[28]。因此,EWAS的一些显著信号可能是由附近的SNP导致的,而SNP或其他影响因素的混淆可能是EWAS鉴定到大量与性状相关的显著DNA甲基化的主要驱动因素。
4 结 论
4.1 本研究对140 头大白猪群体的肉质性状进行 GWAS 分析,鉴定了 516个全基因组水平显著的 SNP 位点以及 68 个位置候选基因,根据候选基因的功能富集分析及相关文献报道筛选了 6 个重要候选基因,分别为 NDUFB6、GALNTL6、IGFBP1、ZFAT、 CDH13 以及 MCUR1。
4.2 本研究通过比较GWAS 和 EWAS 结果鉴定二者重叠的显著关联位点,在b45min关联结果中发现了8个显著CpG位点与 59 个显著SNPs位点位置接近,在DL关联结果中发现1个CpG位点与1个SNP位置接近,在 C22∶6n-3关联结果中发现3个CpG位点与146个显著SNPs位点位置接近。
参考文献(References):
[1] BERRY D P,CONROY S,PABIOU T,et al.Animal breeding strategies can improve meat quality attributes within entire populations[J].Meat Sci,2017,132:6-18.
[2] GRUNDBERG E,MEDURI E,SANDLING J K,et al.Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements[J].Am J Hum Genet,2013,93(5):876-890.
[3] VILLICAA S,BELL J T.Genetic impacts on DNA methylation:research findings and future perspectives[J].Genome Biol,2021,22(1):127.
[4] SCHBELER D.Function and information content of DNA methylation[J].Nature,2015,517(7534):321-326.
[5] BESINGI W,JOHANSSON .Smoke-related DNA methylation changes in the etiology of human disease[J].Hum Mol Genet,2014, 23(9):2290-2297.
[6] 曾浩南,钟展明,徐志婷,等.3款猪50K SNP芯片基因型填充至序列数据的效果评估[J].华南农业大学学报,2022,43(4):10-15.
ZENG H N,ZHONG Z M,XU Z T,et al.Evaluation on genotype imputation performance of three porcine 50K SNP chips from chip data to sequencing data[J].Journal of South China Agricultural University,2022,43(4):10-15.(in Chinese)
[7] CHANG C C,CHOW C C,TELLIER L C,et al.Second-generation PLINK:rising to the challenge of larger and richer datasets[J].GigaScience,2015,4:7.
[8] ZHOU X,STEPHENS M.Genome-wide efficient mixed-model analysis for association studies[J].Nat Genet,2012,44(7): 821-824.
[9] AULCHENKO Y S,RIPKE S,ISAACS A,et al.GenABEL:an R library for genome-wide association analysis[J].Bioinformatics, 2007,23(10):1294-1296.
[10] WANG K,WANG S J,JI X,et al.Epigenome-wide association studies of meat traits in Chinese Yorkshire pigs highlights several DNA methylation loci and genes[J].Front Genet,2023,13:1028711.
[11] AHSAN M,EK W E,RASK-ANDERSEN M,et al.The relative contribution of DNA methylation and genetic variants on protein biomarkers for human diseases[J].PLoS Genet,2017,13(9):e1007005.
[12] MA J,REN J,GUO Y,et al.Genome-wide identification of quantitative trait loci for carcass composition and meat quality in a large-scale White Duroc×Chinese Erhualian resource population[J].Anim Genet,2009,40(5):637-647.
[13] LIU G S,KIM J J,JONAS E,et al.Combined line-cross and half-sib QTL analysis in Duroc-Pietrain population[J].Mamm Genome,2008,19(6):429-438.
[14] PREZ-ENCISO M,CLOP A,NOGUERA J L,et al.A QTL on pig chromosome 4 affects fatty acid metabolism:evidence from an Iberian by Landrace intercross[J].J Anim Sci,2000,78(10):2525-2531.
[15] UEMOTO Y,SOMA Y,SATO S,et al.Genome-wide mapping for fatty acid composition and melting point of fat in a purebred Duroc pig population[J].Anim Genet,2012,43(1):27-34.
[16] PESTA D,JELENIK T,ZAHARIA O P,et al.NDUFB6 polymorphism is associated with physical activity-mediated metabolic changes in type 2 diabetes[J].Front Endocrinol (Lausanne),2021,12:693683.
[17] LING C,POULSEN P,SIMONSSON S,et al.Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle[J].J Clin Invest,2007,117(11):3427-3435.
[18] 罗 培,赵伟杰,王丽娜,等.肌红蛋白和线粒体互作的研究进展[J].中国畜牧杂志,2020,56(5):6-11.
LUO P,ZHAO W J,WANG L N,et al.Advances in myoglobin and mitochondrial interaction[J].Chinese Journal of Animal Science,2020,56(5):6-11.(in Chinese)
[19] MCKEITH R O,KING D A,GRAYSON A L,et al.Mitochondrial abundance and efficiency contribute to lean color of dark cutting beef[J].Meat Sci,2016,116:165-173.
[20] DORAN A G,BERRY D P,CREEVEY C J.Whole genome association study identifies regions of the bovine genome and biological pathways involved in carcass trait performance in Holstein-Friesian cattle[J].BMC Genomics,2014,15(1):837.
[21] GTZ W,DITTJEN O,WICKE M,et al.Immunohistochemical detection of components of the insulin-like growth factor system during skeletal muscle growth in the pig[J].Anat Histol Embryol,2001,30(1):49-56.
[22] DUAN C M,REN H X,GAO S.Insulin-like growth factors (IGFs),IGF receptors,and IGF-binding proteins:roles in skeletal muscle growth and differentiation[J].Gen Comp Endocrinol,2010,167(3):344-351.
[23] ISHIKURA S,NAGAI M,TSUNODA T,et al.The transcriptional regulator Zfat is essential for maintenance and differentiation of the adipocytes[J].J Cell Biochem,2021,122(6):626-638.
[24] 倪和民,赵延辉,邢 凯,等.猪肌内脂肪调控研究进展[J].中国畜牧业,2020(17):91-92.
NI H M, ZHAO Y H, XING K, et al. Research progress on regulation of porcine intramuscular fat[J]. China Animal Industry, 2020(17): 91-92.(in Chinese)
[25] LIU H T,SONG H L,JIANG Y F,et al.A single-step genome wide association study on body size traits using imputation-based whole-genome sequence data in Yorkshire Pigs[J].Front Genet,2021,12:629049.
[26] TOMAR D,DONG Z W,SHANMUGHAPRIYA S,et al.MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics[J].Cell Rep,2016,15(8):1673-1685.
[27] MALLILANKARAMAN K,CRDENAS C,DOONAN P J,et al.MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism[J].Nat Cell Biol,2012,14(12):1336-1343.
[28] VILLICAA S,BELL J T.Genetic impacts on DNA methylation:research findings and future perspectives[J].Genome Biol,2021,22(1):127.
(编辑 郭云雁)
