
枳椇子多糖的酸提取工艺优化及其理化性质与抗氧化活性研究
2024-05-18 01:20:20刘春阳白金波杨尚青史进阳秦亚敏吴德玲解松子
食品与发酵工业 2024年9期
刘春阳,白金波,杨尚青,史进阳,秦亚敏,吴德玲,2,3,解松子,2,3*
1(安徽中医药大学 药学院,安徽 合肥,230012)2(中药研究与开发安徽省重点实验室,安徽 合肥,230012)3(中药饮片制造新技术安徽重点实验室,安徽 合肥,230012)
枳椇子首载于唐代《新修本草》《中华人民共和国卫生部药品标准·中药材》和《香港中药材标准》要求枳椇子的功能性部位为种子[1]。作为一种药食两用中药,枳椇子含有三萜皂苷、黄酮、苯丙素类、多糖等多种化学成分[2],具有保肝、降血糖、抗氧化、降尿酸、调节免疫等药理作用[3]。
植物多糖作为一种生物大分子具有多种生物活性。目前对于枳椇子多糖的研究主要集中在肉质果梗上,而对于种子的研究较为缺乏[4-5]。由于多糖在植物细胞中存在的部位不同,使植物多糖结构复杂,组成多样,多糖的高效提取一直受到广大研究学者的关注,提取方式是影响多糖提取率的重要因素之一。已有研究学者利用响应面法优化枳椇子多糖的水提工艺,使多糖的提取率达到1.84%[6];采用微波辅助技术提取桑叶多糖得到多糖的提取率达到9.41%[7];采用酶辅助法提取黄芪多糖,使多糖得率达到29.96%,高于水提取法[8]。酸提取法是利用稀酸溶液提取多糖的一种方法。已有研究证明,酸提取法能够将含有酸性基团的多糖提取出来,并且能够提高植物多糖的提取率和溶解性;此外,酸提取多糖也具有良好的生物活性,例如,以盐酸为提取溶剂提取玉米须多糖,其得率达到33.36%[9];采用稀盐酸溶液提取得到的羊栖菜多糖具有良好的抗氧化活性;通过比较由不同提取方式得到的海藻多糖的生物活性,发现酸提取多糖能显著性提高与胆汁酸的结合能力[10]。
因此,本文采用酸溶剂提取法提取枳椇子种子部位的粗多糖,经单因素考察和正交试验优化提取工艺参数,通过Sevag法除蛋白、DEAE-52阴离子交换柱对多糖进行分离纯化,并对其进行理化性质分析和体外抗氧化活性评价,以期为枳椇子多糖的综合开发利用提供理论依据。
1 材料与方法
1.1 材料与试剂
枳椇子,安徽亳州,烘干粉碎后备用;葡聚糖分子质量标准品,上海源叶生物科技有限公司;DEAE纤维素DE-52,北京索莱宝科技有限公司;DPPH、ABTS试剂,上海阿拉丁生化科技股份有限公司。
1.2 仪器与设备
FD-1A-50冷冻干燥机,博医康(北京)仪器有限公司;HC-3018高速离心机,安徽中科中佳科学仪器有限公司;SPECORD S600紫外可见分光光度计,上海元析仪器有限公司;RID-10A凝胶色谱仪,岛津;DHL-A电脑数显恒流泵,上海沪西分析仪器厂有限公司;SpectraMax i3X酶标仪,赛默飞世尔科技公司;尼力高6700傅立叶交换显微红外成像光谱仪,美国尼高力仪器公司。
1.3 实验方法
1.3.1 实验样品预处理
枳椇子原药材洗净、烘干、打粉,称取适量药材粉末以石油醚为溶剂脱脂,脱脂后的滤渣晾干备用。取80%(体积分数)的乙醇与该滤渣混匀,80 ℃回流脱单糖,滤渣烘干备用。
1.3.2 枳椇子多糖提取
称取适量的枳椇子粉末,加入适量的HCl溶液进行提取,提取液过滤,滤液立即用0.5 mol/L NaOH溶液中和,5 000 r/min离心10 min后去除沉淀,浓缩至一定体积,加入无水乙醇使提取液中乙醇体积分数为80%,静置24 h,所得醇沉物复溶后加Sevag试剂搅拌离心,直至无蛋白沉淀产生。糖液除去残留的有机试剂后,蒸馏水透析,冷冻干燥。
1.3.3 枳椇子多糖提取率和含量测定
标准曲线的绘制:称取适量的无水葡萄糖,配成0.1 mg/mL的葡萄糖标准溶液,分别取0、0.05、0.10、0.15、0.20、0.25、0.30、0.35 mL标准溶液于试管中,蒸馏水补足至1 mL。加入2.5 μL 80%苯酚溶液(体积分数,下同),再加入2.5 mL浓硫酸,混匀后室温反应20 min,用紫外可见分光光度计在490 nm处检测吸光度值。以无水葡萄糖浓度为横坐标,吸光度值为纵坐标绘制标准曲线:y=21.45x-0.008 1,R2=0.990 2。
取0.1 mg/mL的多糖样品溶液1 mL,加入25 μL 80%苯酚溶液,再加入2.5 mL浓硫酸,涡旋混匀后室温反应20 min后,用紫外分光光度计在490 nm处检测吸光度值,枳椇子粗多糖得率的计算如公式(1)所示:
(1)
式中:m1,枳椇子多糖的质量,mg;m2,样品质量,mg。
1.3.4 单因素试验
以枳椇子原药材粉末为原料,固定提取温度(A)80 ℃、料液(B)为1∶60(g∶mL)、提取时间(C)2 h、盐酸浓度(D)0.2 mol/L,改变某一因素探讨对枳椇子多糖得率的影响,单因素设计如表1所示。

表1 单因素试验设计Table 1 Single factor experiment design
1.3.5 正交试验
根据单因素试验结果,采用四因素三水平正交试验进行提取工艺优化。正交试验设计见表2,每组平行做3次,多糖得率结果取均值并验证其合理性及稳定性。

表2 酸提取法正交试验因素与水平Table 2 Factors and levels of orthogonal experiment of acid extraction method
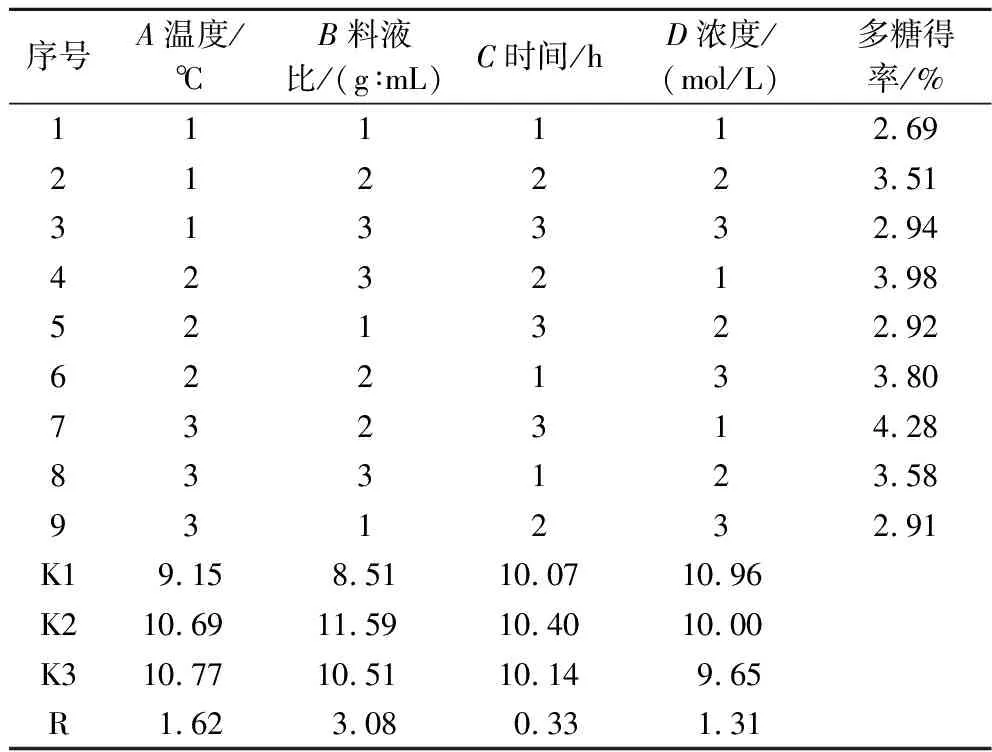
表3 酸提取法正交试验结果Table 3 Orthogonal experiment results of acid extraction method
1.3.6 分离纯化
将冻干的HDP用超纯水配制为10 mg/mL的溶液,过滤,加入预先用Tris-HCl平衡的DEAE-52纤维素阴离子交换柱(1.6 cm×60 cm)中,以5.0 mL/min的流速分别用超纯水和不同梯度浓度的NaCl溶液(0.1、0.2、0.3 mol/L)洗脱。收集同一洗脱液的糖溶液透析、浓缩、冷冻干燥后备用。
1.3.7 化学成分测定
蛋白质含量测定采用考马斯亮蓝法[11],根据回归方程y=7.735 7x-0.002 6(R2=0.994 8)计算;糖醛酸含量测定采用间羟基联苯法[12],根据回归方程y=4.672 6x-0.028 9(R2=0.997 5)计算。
1.3.8 结构鉴定
1.3.8.1 分子质量测定
采用高效凝胶渗透色谱法测定枳椇子多糖的分子质量,以不同分子质量的葡聚糖标准品保留时间绘制标准曲线,色谱条件:凝胶柱为TSK G4000PWxl色谱柱,流动相为过膜双蒸水,流速0.6 mL/min,进样量20 μL,柱温30 ℃,示差折光检测器[13]。
1.3.8.2 单糖组成
参考张璐等[14]的方法采用高效液相色谱法测定HDP中的单糖组成。
1.3.8.3 紫外全波长扫描
取适量枳椇子多糖溶于双蒸水中,紫外分光光度计在190~800 nm范围内进行扫描。
1.3.8.4 红外光谱
称取2 mg充分干燥后的枳椇子多糖,与200 mg充分干燥的KBr压片,红外光谱扫描仪上4 000~400 cm-1范围内对压片进行扫描分析。
1.3.8.5 刚果红实验
取2 mL 2.5 mg/mL多糖溶液与2 mL刚果红溶液置于试管中混匀,再向混合溶液中依次添加不同体积的NaOH溶液使混合溶液中NaOH浓度为0、0.05、0.10、0.15、0.20、0.25、0.30、0.35、0.40、0.45、0.50 mol/L。并以不含枳椇子多糖溶液的混合溶液(蒸馏水与刚果红混合溶液)作为对照。在室温下静置10 min后,测定不同浓度NaOH混合溶液的最大吸收值。
1.3.8.6 碘-碘化钾实验
配制1 mg/mL的枳椇子多糖溶液,加入1.2 mL碘试剂,混匀后用紫外分光光度计对在300~700 nm范围内进行扫描。
1.3.8.7 扫描电镜分析
称取少量枳椇子多糖,均匀涂置于带有导电胶的样品台,使用扫描电镜高真空观察枳椇子多糖表观特征。
1.3.9 体外抗氧化活性
1.3.9.1 DPPH自由基清除能力测定
参考滕欢欢等[15]的方法并稍作调整,配制浓度为0.2 mmol/L的DPPH乙醇溶液和不同质量浓度(0.2、0.4、0.6、0.8、1.0、2.0、3.0 mg/mL)枳椇子多糖溶液。取试管依次加入2 mL DPPH溶液后再加入2 mL不同浓度的枳椇子多糖溶液摇匀,避光反应30 min,测定其在517 nm处的吸光度值。抗坏血酸(维生素C)为阳性对照,蒸馏水为空白对照,计算清除率。平行测定3次,取平均值,其计算如公式(2)所示:
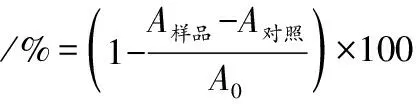
(2)
式中:A样品,样品与DPPH混合溶液OD值;A0,去离子水同DPPH混合溶液OD值;A对照,样品同去离子水混合溶液OD值。
1.3.9.2 羟自由基(·OH)清除能力测定
参考LI等[16]的方法并稍作调整,配制0.2、0.4、0.6、0.8、1.0、2.0、3.0 mol/L的枳椇子多糖溶液。取7只试管,分别加入上述不同的枳椇子多糖溶液1 mL,随后向试管中依次加入6 mmol/L的FeSO4溶液1 mL,再分别加入6 mmol/L的H2O2溶液1 mL,摇匀,静置10 min,再加入6 mmol/L的水杨酸溶液1 mL,摇匀,静置30 min后,于510 nm处测其吸光值。以抗坏血酸(维生素C)作为阳性对照。其计算如公式(3)所示:
(3)
式中:A样品,样品与反应溶液OD值;A对照,样品与蒸馏水混合OD值;A0,蒸馏水与反应溶液OD值。
1.3.9.3 ABTS阳离子自由基清除能力测定
参考XIE等[17]的方法并稍作调整,配制0.2、0.4、0.6、0.8、1.0、2.0、3.0 mol/L枳椇子多糖溶液,分别取1 mL不同浓度多糖溶液与预先配制好的ABTS工作液2 mL混匀,避光1 h后在734 nm处测定吸光度值。以抗坏血酸(维生素C)作为阳性对照。其计算如公式(4)所示:
(4)
式中:A样品,样品与ABTS反应溶液OD值;A对照,样品与PBS混合OD值;A0,蒸馏水与ABTS反应溶液混合OD值。
1.4 数据处理
采用SPSS 23.0进行正交试验设计和单因素方差分析,图采用Origin 2021和GraphPad Prism 8.2.1绘制。上述实验均重复3次,实验结果采用平均值±标准差的形式表示。
2 结果与分析
2.1 单因素试验结果
2.1.1 料液比对提取率的影响
料液比是影响多糖提取率的重要因素之一,合适的料液比能通过改变细胞内外渗透压而使多糖充分溶出[18]。如图1所示,枳椇子多糖得率随着料液比的增大而出现先上升后下降的趋势,随着料液比的等比提高,溶液黏度降低,分子扩散速度加快,多糖溶出率提高,拐点出现在1∶100,此时提取率为(3.7±0.07)%。在1∶100之后多糖得率有所下降,当料液比为1∶140时,多糖提取率为(2.2±0.14)%。这可能是由于此时组织内外浓度差消失,多糖溶出率不再变化,酸溶液使多糖发生部分降解,造成提取率下降。故确定料液比(g∶mL)在正交优化中的因素水平为1∶80、1∶100、1∶120。
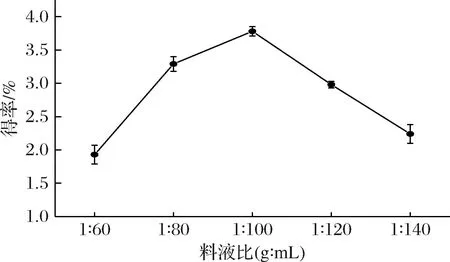
图1 不同料液比对枳椇子多糖得率的影响Fig.1 Effect of solid-liquid on polysaccharide yield of H. dulcis
2.1.2 盐酸浓度对提取率的影响
如图2所示,多糖得率随着盐酸浓度提高而先增后减,在0.2~0.4 mol/L这一区间多糖得率增加不明显,0.1~0.2 mol/L和0.4~0.5 mol/L出现急剧的上升和下降趋势,前者可能是由于酸溶液对药材细胞壁的破坏使得多糖大量溶出,后者可能是由于高浓度的酸造成多糖降解,造成多糖损失[9]。故确定盐酸浓度在正交优化中的因素水平为0.2、0.3、0.4 mol/L。
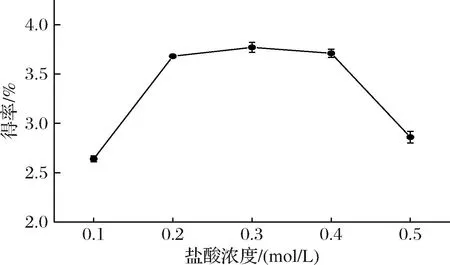
图2 不同提取浓度对枳椇子多糖得率的影响Fig.2 Effect of extract concentration on polysaccharide yield of H. dulcis
2.1.3 时间对提取率的影响
提取时间是影响多糖得率的又一重要因素。如图3所示,多糖得率随着提取时间的等时延长而出现先升高后下降的趋势。其拐点是2.5 h,提取率为(4.1±0.09)%。1 h和1.5 h的多糖得率分别是(3.0±0.06)%和(3.6±0.15)%,这可能是因为提取时间较短,使得多糖不能充分溶出;在提取时间为3 h时多糖得率为(3.9±0.03)%,呈现缓慢下降的趋势,这可能是因为长时间浸提导致多糖发生降解,从而影响多糖的提取率[19]。故确定提取时间在正交优化中的因素水平为2、2.5、3 h。
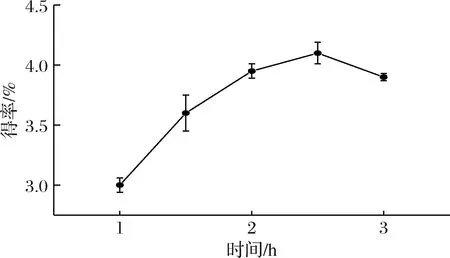
图3 不同提取时间对枳椇子多糖得率的影响Fig.3 Effect of extract time on polysaccharide yield of H. dulcis
2.1.4 温度对提取率的影响
如图4所示,多糖得率随着温度升高出现先上升后下降的趋势。这与分子间的运动有关,随着温度的升高分子间运动速度加快,多糖溶出率增加,拐点出现在80 ℃,此时得率达到(3.9±0.19)%,但这种溶出是有限度的,当温度达到100 ℃时,多糖得率下降到(3.4±0.21)%,这可能因为在温度过高的情况下多糖结构会受到破坏,影响多糖得率[20]。故确定温度在正交优化中的因素水平为70、80、90 ℃。

图4 不同提取温度对枳椇子多糖得率的影响Fig.4 Effect of extract temperature on polysaccharide yield of H. dulcis

图5 枳椇子多糖DEAE洗脱曲线Fig.5 Elution curve of polysaccharide of H. dulcis
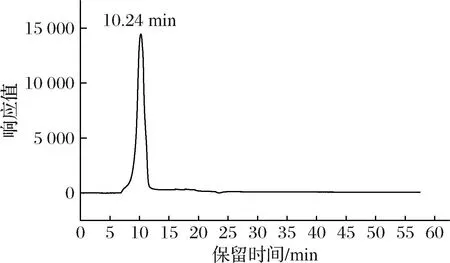
图6 分子质量分布Fig.6 HPGPC profile of molecular weight distribution
2.2 正交试验结果
通过直观分析R值得出4个因素对枳椇子多糖提取的影响大小次序为料液比>温度>浓度>时间。因C组极值很小,所以将此列作为方差分析的误差列,结果见表4,根据方差分析结果可知,因素A、因素B,均具有显著性差异,P<0.05;而因素C及因素D均没有显著性。

表4 正交试验方差分析Table 4 Analysis of variance of orthogonal test
由正交结果分析得出酸提枳椇子多糖的最佳工艺组合为A3B2C3D1,提取条件为温度90 ℃,料液比为1∶100(g∶mL),时间为3 h,浓度为0.2 mol/L。
按照最优工艺组合A3B2C3D1提取,并计算多糖得率。结果如表5所示,在最佳工艺条件下,枳椇子多糖得率均值为(4.07±0.07)%,相对标准偏差(relative standard deviation, RSD)为1.69%,说明优选出的最佳提取工艺合理且稳定,并且高于于刚等[21]水提枳椇子多糖的提取率。由表3可知,因素C的R值为0.33,极差较小,说明提取时间对该提取工艺的影响不大,考虑到节约能耗,故将正交最佳工艺调整为A3B2C2D1,即温度90 ℃,料液比为1∶100(g∶mL),时间为2.5 h,盐酸浓度为0.2 mol/L。

表5 正交试验验证结果Table 5 Orthogonal experiment verification results
2.3 枳椇子多糖分离纯化
枳椇子粗多糖溶液经DEAE-52纤维素离子交换色谱柱分离,依次用超纯水和0.1、0.2、0.3 mol/L的NaCl溶液梯度洗脱,依次得到4个组分,其得率分别是6.7%、7.0%、8.5%、26.7%。其中0.3 mol/L盐洗组分得率最高,因此对该组分进行后续理化性质和生物活性研究,命名为HDP。
2.4 HDP的化学成分分析
HDP的碳水化合物含量占(67.86±0.19)%,蛋白质含量占(4.07±0.03)%,糖醛酸含量占(41.57±0.36)%,经该方法提取纯化得到的枳椇子种子部位的多糖含量低于蔡冰洁等[22]提取纯化的果梗部位的多糖含量,但种子部位多糖的糖醛酸含量高于果梗部位,推断HDP为酸性多糖。
2.5 HDP分子质量
如图2所示,HPD的出峰时间为10.24 min,为单一对称峰,说明HDP经分离纯化后为均一性多糖。根据多糖分子质量标准曲线Y=-2.052 2x+24.945计算,计算得出HDP的分子质量为3.03×105Da。强明亮[23]分离纯化得到枳椇果梗部位的多糖,分子质量为3.65×105Da,杨兵[24]从果梗部位得到一个均一性多糖,分子质量为3.73×105Da,而对于同为鼠李科的骏枣多糖,其分子质量为3.24×104Da[25]。这说明处于同一科的植物多糖其分子质量可能存在较大差异,而对于同一属的植物中多糖的分子质量差异相对较小。
2.6 HDP单糖组成分析
如图7所示,HDP由甘露糖、氨基葡萄糖、鼠李糖、葡萄糖醛酸、半乳糖醛酸、葡萄糖、半乳糖、鼠李糖、阿拉伯糖组成,摩尔百分比分别是10.46%、1.44%、8.19%、9.79%、4.54%、4.61%、38.02%、9.95%、13.01%。主要单糖组成包括半乳糖、阿拉伯糖、甘露糖和葡萄糖醛酸。有研究报道,枳椇果梗多糖由甘露糖、鼠李糖、葡萄糖醛酸、半乳糖醛酸、葡萄糖、半乳糖和阿拉伯糖组成,各单糖的摩尔百分比分别是3.64%、1.41%、4.67%、5.16%、3.01%、60.02%、22.09%,半乳糖和阿拉伯糖是果梗多糖的主要单糖组分[24]。单糖组成结果证实HDP为酸性多糖,HDP的单糖组成与已发表文献所报道的枳椇果梗多糖的单糖组成存在类似性,但单糖的组成比例有显著差异,这可能与提取的部位和提取方法有关[22]。
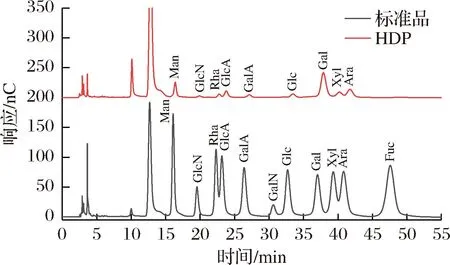
图7 单糖组成Fig.7 Monosaccharide composition of HDP
2.7 HDP的紫外光谱和红外光谱分析
核酸和蛋白质在260 nm和280 nm附近有吸收峰,可以利用紫外全波长扫描来检测多糖中是否含有游离的蛋白质和核酸。由图8-a可知,HDP在260 nm处无吸收峰,在280 nm处有微弱吸收峰,说明HDP不含核酸,有少量蛋白存在。
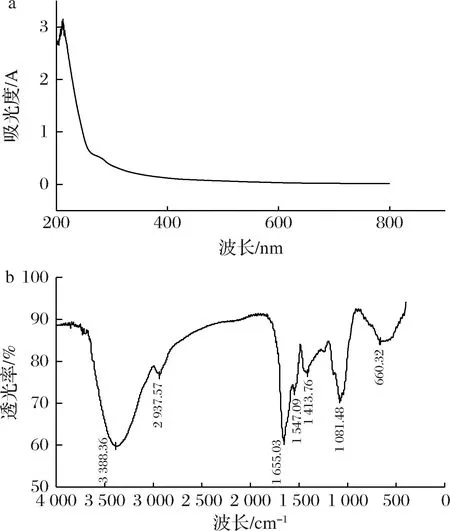
a-紫外光谱;b-红外光谱

2.8 HDP构象分析
刚果红试剂可与三股螺旋结构的多糖形成复合物,复合物未接触碱液时紫外扫描会出现红移,接触碱液后随着碱液浓度增加而逐渐蓝移至稳定[29]。由图9-a可知,与对照组不同,HDP与刚果红混合溶液未出现先红移后蓝移最后趋于稳定的现象,因此HDP没有规则螺旋构象,这可能与HDP较复杂的单糖组成有关[30];由图9-b可知,HDP在350 nm处有强吸收峰,而在565 nm左右没有吸收峰。说明HDP主干上具有复杂的链状结构,侧链较长,支链较多。

a-三股螺旋结构;b-碘-碘化钾紫外扫描图

a-550×;b-2 000×;c-3 000×
2.9 HDP微观表面形态分析
为了进一步研究枳椇子多糖的微观结构,采用扫描电镜观察HDP表面微观形态,HDP呈不规则层片状分布,表面光滑致密,这可能与HDP中的阿拉伯糖与带电基团形成氢键有关[31];在高倍镜下,HDP表面出现空洞,并伴有少量裂痕,边缘有不规则层片状堆叠,这可能是由于酸提取溶剂在一定程度上影响HDP的表面微观结构。
2.10 HDP抗氧化活性评价
过量活性氧诱导的氧化应激反应会对细胞结构和生物分子的功能造成严重损伤,从而直接或间接地引发衰老、炎症和心血管疾病[32]。DPPH自由基较为稳定,抗氧化剂能够将稳定的DPPH还原成非自由基形式的DPPH-H,从而使紫红色的溶液迅速褪色。ABTS阳离子自由基是一种呈蓝绿色的自由基,当与抗氧化剂结合后能使溶液褪色以此评价多糖的自由基清除能力。·OH是活性氧中最强的一种,能快速穿透细胞膜与蛋白质、核酸等重要的生物大分子发生反应导致组织损伤和细胞死亡,因此·OH的清除对机体保护十分重要[33]。
HDP的自由基清除能力测定结果如图11所示,DPPH自由基、ABTS阳离子和·OH清除率随着多糖浓度的升高而增强,在多糖质量浓度为3.0 mg/mL时,DPPH自由基、ABTS阳离子和·OH清除率分别达到(61.67±0.15)%、(83.88±0.14)%、(58.89±0.91)%。经计算,HDP的IC50分别是1.178、0.867、2.112 mg/mL,表现出良好的体外抗氧化能力,但枳椇子多糖的抗氧化能力劣于拐枣多糖[34](在0.1 mg/mL时,对DPPH的清除率为86.73%;在2 mg/mL时,对ABTS的清除率为91.33%)。
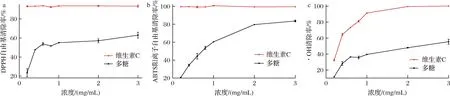
a-DPPH自由基清除率;b-ABTS阳离子自由基清除率;c-·OH清除率
理化性质是影响植物多糖抗氧化能力的关键因素。有研究报道,多糖清除自由基的能力通常取决于其供电子或供氢的能力,多糖的羧基可以与异碳的氢原子发生反应[35],因此较高的糖醛酸含量有利于提高多糖的自由基清除能力,本实验得到HDP含有41.57%的糖醛酸,红外光谱和单糖组成的结果也证实糖醛酸的存在,HDP表现较好的清除自由基能力可能与糖醛酸的含量有很大关系。HDP的分子质量为3.03×105Da,主要由半乳糖、阿拉伯糖、甘露糖组成。多糖的分子质量也是影响多糖抗氧化能力的关键因素,分子质量过大不能透过细胞膜,分子质量过小则不能形成“活性中心”[24],有学者通过降解得到分子质量在6.60×105~8.80×105Da范围的多糖,发现它们具有良好的抗氧化能力,这可能与较低分子质量的多糖具有更多的还原羟基,从而能与更多的自由基结合有关[36]。也有报道称,葡萄糖占比少的多糖具有更好的抗氧化能力[37];此外,有研究证实以半乳糖为主链,阿拉伯糖为侧链的阿拉伯半乳聚糖能表现出良好的抗氧化和抗衰老能力[38]。因此,HDP良好的抗氧化能力可能是糖醛酸含量、分子质量大小、单糖组成等多个理化性质共同作用的结果。
3 结论
本文利用单因素考察和正交试验探究酸提枳椇子多糖的提取工艺,得到最优提取条件为温度90 ℃,液料比1∶100(g∶mL),提取时间为2.5 h,盐酸浓度为0.2 mol/L。在此工艺下多糖得率达到4.07%。通过对枳椇子粗多糖进行纯化,以得率最高的HDP为对象进行理化性质分析和抗氧化活性评价。经测定HDP分子质量为3.03×105Da,含有甘露糖、氨基葡萄糖、鼠李糖、葡萄糖醛酸、半乳糖醛酸、葡萄糖、半乳糖、鼠李糖、阿拉伯糖,是一种吡喃环型酸性多糖,具有无规则螺旋结构并含有较多分支,并表现出层片状的微观形态。HDP能够有效清除ABTS阳离子自由基、DPPH自由基、羟自由基,表现出良好的体外抗氧化活力。本文主要研究了酸提枳椇子多糖的提取工艺、理化性质以及体外抗氧化活性。此后还需要进一步对枳椇子多糖进行深入的结构表征和体内生物活性研究,该研究将为枳椇子功能性食品的开发提供理论基础。
猜你喜欢
中成药(2021年5期)2021-07-21 08:38:18
当代水产(2020年4期)2020-06-16 03:23:22
当代水产(2020年3期)2020-06-15 12:02:52
心声歌刊(2019年1期)2019-05-09 03:21:30
天然产物研究与开发(2019年1期)2019-03-01 05:41:20
科学中国人(2018年8期)2018-07-23 02:26:46
饮食与健康·下旬刊(2017年3期)2017-03-30 21:36:44
饮食科学(2016年10期)2016-11-19 09:42:47
天然产物研究与开发(2016年1期)2016-06-05 10:29:25
中南民族大学学报(自然科学版)(2015年2期)2015-12-16 12:11:10
