
AB-8树脂联用C18柱分离牡丹籽粕中新橙皮苷及生物活性研究
2024-05-18 01:20邹平陈文涛胡建刚徐莹张迎阳孙承骏夏炜芳高蕙文
食品与发酵工业 2024年9期
邹平,陈文涛,胡建刚,徐莹,张迎阳,孙承骏,夏炜芳,高蕙文
1(常州大学 药学院/生物与食品工程学院,江苏 常州,213164)2(绍兴市食品药品检验研究院,浙江 绍兴,312071)3(常州市食品药品纤维质量监督检验中心,江苏 常州,213022)
牡丹是中国特有的植物[1],具有良好的观赏和药用价值,是一种重要的经济作物,具有数千年的生长和栽培历史。油牡丹是一种牡丹品种,牡丹籽可用于制备食用油。牡丹籽粕为牡丹籽榨油后的副产品,目前的研究表明,已经从牡丹籽粕提取了许多种类的活性成分,包括黄酮类化合物[2],多酚[3],功能性多糖[4]等。许多报告表明,类黄酮具有许多有益的生物学特性[5-6],例如抗氧化、抗炎、抗病毒和抗肿瘤特性。新橙皮苷是一类纯天然的类黄酮物质,有着强大的抗氧化和抗炎作用,它能够诱导MDA-MB-231细胞凋亡,并且在DPPH自由基消除实验中显示出显著的抗氧化活性[7]。新橙皮苷可以显著促进胃分泌物和胃酸产生量,从而增加服用葡萄糖耐量和胰岛素敏感度,并且明显降低血糖小鼠的胰岛素抵抗,并且有效降低血清甘油三酯、总胆固醇、瘦素水平和肝功能指标,从而达到治疗疾病的目的[8]。CHEN等[9]建立了一种利用高效液相紫外光谱(HPLC-ultra-violet spectroscopy, HPLC-UV)分离定量新橙皮苷的方法,新橙皮苷的检测限(limit of detection, LOD)和定量限(limit of quantitation, LOQ)分别低于0.84 mg/mL和2.84 mg/mL;UCHIYAMA等[10]通过正相HPLC分离商业样品中的新橙皮苷,相对比例为84%;WANG等[11]构建了一种新型乙醇两相系统(ethanolic two-phase system, ETPS),用于从柚子中分离新橙皮苷,回收率为68.03%;DALMAU等[12]利用超声辅助的方法从橙副产物中提取新橙皮苷,提取率为39%。新橙皮苷作为黄酮类化合物的一种,具有较强的生物活性,WANG等[13]评估了新橙皮苷的抗氧化活性,发现新橙皮苷通过激活人骨肉瘤细胞中的ROS/JNK通路引起G2/M期阻滞并诱导细胞凋亡和自噬;ORTIZ等[14]研究发现新橙皮苷具有抗破骨细胞和抗炎作用,抑制破骨细胞标志物的表达,降低活性氧、促炎细胞因子和基质金属蛋白酶水平;ZHANG等[15]证实了新橙皮苷在影响细胞凋亡、细胞生长、肿瘤发生和肿瘤微环境中起着重要作用。
本实验通过前期探索,利用有机溶剂提取牡丹籽粕中的黄酮类化合物(flavonoids in peony seed meal,PMSF),借助吸附动力学筛选出AB-8大孔树脂与C18柱联用分离新橙皮苷的方法,同时探究了新橙皮苷的生物活性(抗氧化、抑制癌细胞增殖),分子动力学模型揭示了其抑制癌细胞增殖的作用机理,为进一步开发牡丹籽粕产品提供理论依据。
1 材料与方法
1.1 试剂与仪器
油牡丹籽粕,中国江苏国色天香油用牡丹科技发展有限公司;芦丁、无水乙醇、乙腈、三氟乙酸、DPPH、ABTS、氯化硝基四氮唑蓝(nitrotetrazolium chloride,NBT)、吩嗪硫酸甲酯(phenazine methosulfate,PMS)、NADH(均为分析纯),阿拉丁(上海)生物试剂公司;C18球形硅胶柱,常州三泰生物科技公司;大孔树脂AB-8、X-5、DM301、CAD-40、D101、NKA,广州伟伯科技有限公司。
721 N可见光分光光度计,上海仪电分析仪器有限公司;多功能离心机,德国Eppendorf公司;SHB-ⅢA环水式真空泵,上海聚昆仪器设备有限公司;ATY224精密电子天平,常州万泰天平仪器有限公司;UV-3600紫外可见近红外分光光度计,岛津企业管理(中国)有限公司;Q ExactiveTMPlus 质谱仪,赛默飞世尔科技公司。
1.2 实验方法
1.2.1 PMSF的处理
1.2.1.1 PMSF的提取
将牡丹籽粕经过粉碎机破碎后,筛选出5.00 g粉末,加入乙醇溶剂,放入恒温磁力搅拌机中。根据实验室前期实验[16]结果可知PSMF提取的最佳条件为料液比1∶15,浸提温度50 ℃,乙醇体积分数70%,浸提时间30 min,此时PSMF质量浓度最高,为(234.84±1.17) mg/mL。
1.2.1.2 标准曲线的绘制
PSMF含量的测定方法根据文献[16]修改如下:取10 mg芦丁标准品配制50 mg/mL对照液,稀释成0.0~50.0 mg/mL不同质量浓度梯度,再加入5 mL 10 g/L AlCl3溶液,摇匀后静置10 min,以吸光度(y)为纵坐标,芦丁含量(x)为横坐标在415 nm处测定吸光度并绘制标准曲线图。芦丁对照品溶液在0.00~50.00 mg/mL,吸光度与芦丁的浓度呈良好的线性关系,回归方程为:y=(0.03±2.01E-4)x+(0.01±0.006),R2=0.999 45。
1.2.2 大孔树脂的筛选
1.2.2.1 大孔树脂预处理
本实验选取了6种不同性质(弱极性、中极性和非极性)的大孔树脂,它们是AB-8、X-5、DM301、CAD-40、D101和NKA。为了确保大孔树脂的分离性质[17],先用95%乙醇(体积分数)浸泡24 h,接着用蒸馏水多次冲刷,直到没有明显的酒精味,流出液无浑浊后。最后,用40 g/L NaOH浸泡24 h,以除去树脂合成过程中的杂物,以免对样品吸收产生负面影响。将处理过的树脂用蒸馏水彻底洗净,之后用双层滤纸过滤,以备后续使用。
1.2.2.2 大孔树脂的参数
不同性质的大孔树脂对于不同极性的物质具有不同分离效果,综合比较6种不同大孔树脂的吸附率、解吸率的差异来筛选树脂。不同树脂参数见表1。
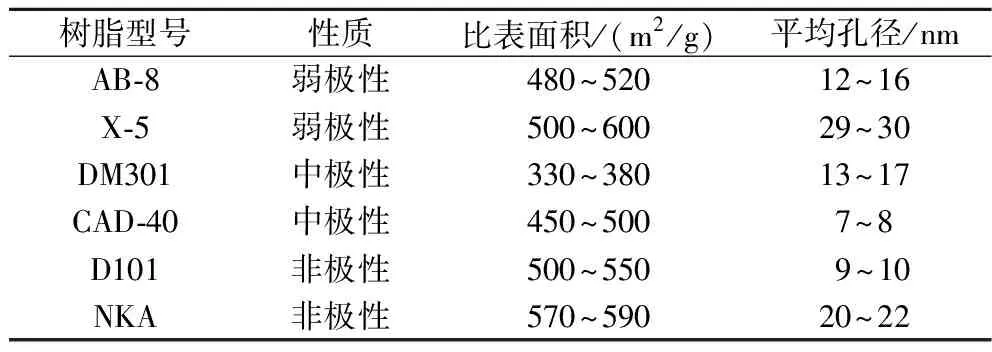
表1 六种不同大孔树脂的性质比较Table 1 Comparison of properties of six different macroporous resins
1.2.2.3 吸附率的计算
从AB-8、X-5、DM301、CAD-40、D101和NKA共6种大孔树脂溶液中取出5 g,放入锥形瓶内,加入20 mL PMSF溶液,恒温振荡12 h(30 ℃,120 r/min),取出上清液,计算溶液中剩余PMSF浓度。吸附速率的计算如公式(1)所示:
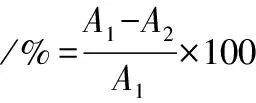
(1)
1.2.2.4 解吸率的计算
将大孔树脂溶液放入锥形瓶内,加入20 mL 75%乙醇,并将其放入恒温振荡箱中,在30 ℃、120 r/min的恒温条件下,充分吸收12 h,取上清液测定溶液中剩余PMSF含量,这是因为乙醇可以与大孔树脂溶液中的分子态产物竞争吸附,使产物溶解于乙醇,从而从大孔树脂溶液上解析出PMSF。解吸率的计算如公式(2)所示:
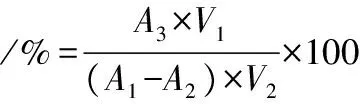
(2)
式中:A1,吸附前溶液中PMSF含量,mg/mL;A2,吸附后溶液中剩余PMSF含量,mg/mL;A3,解吸后溶液中PMSF含量,mg/mL;V1,解吸液体积,mL;V2,吸附液体积,mL。
1.2.2.5 吸附动力学
吸附动力学研究在振荡培养箱中以25 r/min的速度在30 ℃下进行。分别在0~120 min每10 min对来自6种不同树脂样品进行紫外分析。以上述清液中PMSF的浓度为横坐标,以吸附平衡时的吸附量为纵坐标,绘制出树脂准一级动力学模型和准二级动力学模型的等温吸附曲线[18]。同时,利用Langmuir和Freundlich吸附模型[19],得到了相应的等温吸附方程。其计算如公式(3)和公式(4)所示:
准一级动力学方程为:
ln(Qe-Qt)=lnQe-K1×t
(3)
准二级动力学方程为:
此外,TGF-β还可抑制MMPs的活性以及诱导蛋白酶抑制剂如纤溶酶原激活物抑制剂1 (plasminogen activator inhibitor 1,PAI-1)和金属蛋白酶组织抑制物(tissue inhibitor of metalloproteinases,TIMPs)的合成,发挥基质保存作用[24-25]。
(4)
式中:Qe,树脂在PMSF吸收稳定时的吸收量,mg/g;Qt,t时刻树脂的吸收量,mg/g;K1和K2,动力学参数。
Langmuir等温吸附如公式(5)所示:
(5)
Freundlich等温吸附如公式(6)所示:
(6)
式中:Ce,吸收完全后溶液中PMSF的含量,mg/L;Qe,吸收稳定时的吸收量,mg/L;Qm,最大吸附量,mg/L;Ka,Langmuir方程式中大孔树脂与PMSF之间推动力的大小程度;Kb,Freundlich方程式中与大孔树脂吸附量有关的系数。
1.2.3 动态洗脱工艺的确定
1.2.3.1 确定最佳上样流速
选用30 mm×300 mm的层析柱,将层析柱垂直安装,采用湿法装柱,将大孔树脂装入层析柱后用蒸馏水冲洗平衡,然后上样10 mL 1 mg/mL的PMSF液。实验选择了2、4、6、8、10 BV/h的流速进行收集。在大孔树脂吸附30 min,加入75%乙醇,每管收集1 mL。然后,按照1.2.1.2节的方法计算了PMSF含量,并绘制曲线,确定最佳上样流速。
1.2.3.2 确定最佳上样量
采用最佳上样速率开始采集,每管采集1 mL,测量PMSF含量,描绘泄漏曲线,确定最佳上样量。
1.2.3.3 确定最佳乙醇洗脱浓度
通过控制最佳上样量和流速,以及使用不同浓度的乙醇加以洗脱,每管收集1 mL,计算PMSF含量,描绘曲线,确定最佳乙醇洗脱浓度。
1.2.3.4 确定最佳洗脱速度
实验洗脱流速分别为1、2、3、4、5、6 mL/min。每管收集1 mL,测定PMSF含量,绘制曲线,确定最佳洗脱速度。
1.2.3.5 大孔树脂分离
根据最佳上样量、最佳上样流速、最佳乙醇洗脱浓度、最佳洗脱速度进行吸附分离,分离后的不同组分测试其抗氧化能力。
1.2.4 C18柱分离
C18球形硅胶柱选用常州三泰科技公司分离柱,粒径15 mm,孔径100 Å,具体信息见表2。流动相选用含有0.1%三氟乙酸的70%乙腈-水溶液(体积分数),选用经大孔树脂分离后的组分上样,上样量为2 mL,流速1 mL/min。
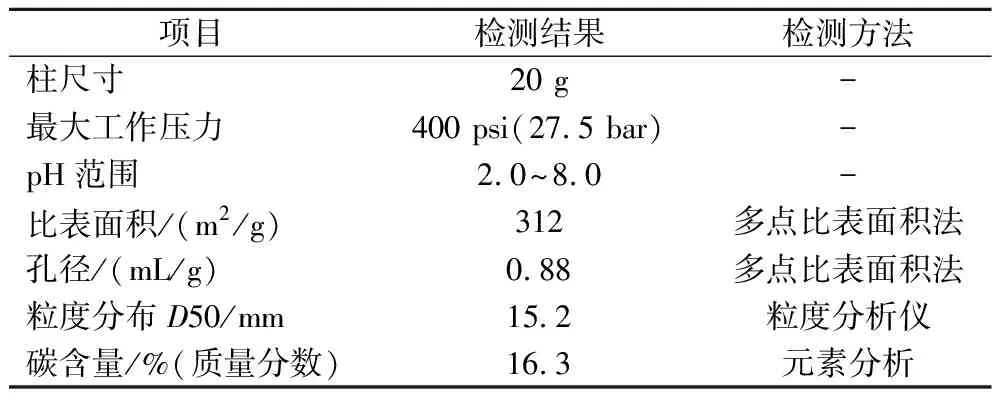
表2 C18球形硅胶柱性质Table 2 Properties of C18 spherical silica gel column
1.2.5 抗氧化活性
1.2.5.1 DPPH自由基清除率
根据文献[20],稍加修改。分别配制成质量浓度(200、400、600、800、1 000 mg/mL)的PMSF。将1 mL 0.2 mmol/L DPPH自由基溶液(95%乙醇溶解)加入3 mL样品,避光反应30 min,517 nm处测定了样品的吸光度。空白为1 mL样品+3 mL 95%乙醇,对照为1 mL DPPH+3 mL 95%乙醇溶液。在相同条件下测定A517。DPPH自由基清除率的计算如公式(7)所示:
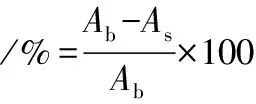
(7)
式中:As、Ab,样品和空白的吸光度。
1.2.5.2 超氧阴离子自由基清除率
参照文献[20]并稍加修改。具体方法如下,分别配制成质量浓度(200、400、600、800、1 000 mg/mL)的PMSF。1.5 mL PMSF液,依次加入0.5 mL 300 mmol/L NBT(pH 8.0 Tris-HCl缓冲液配制),0.5 mL 468 mmol/L NADH(pH 8.0 Tris-HCl缓冲液配制),0.5 mL 60 mmol/L PMS(pH 8.0 Tris-HCl缓冲液配制),冲击混合,25 ℃水浴5 min,560 nm处测定光吸收值,以缓冲液代替样品作为空白对照。超氧阴离子自由基清除率的计算如公式(8)所示:
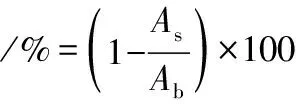
(8)
式中:As、Ab,样品和空白吸光度。
1.2.5.3 ABTS阳离子自由基清除率
根据文献[20]的方法进行。将5 mL 7 mmol/L ABTS溶液与88 mL 140 mmol/L过硫酸钾溶液混合,在20 ℃下放置20 h,加入约3倍75%乙醇,在734 nm处的吸光度为0.70±0.02,得到ABTS阳离子溶液。分别配制成质量浓度200、400、600、800、1 000 mg/mL的PMSF。样品由9.8 mL稀释的ABTS阳离子溶液和0.2 mL PMSF组成。以0.2 mL蒸馏水和9.8 mL稀释的ABTS阳离子溶液的混合物为空白,以0.2 mL PMSF和9.8 mL蒸馏水为对照。所有混合物在室温下放置0.5 h,用分光光度计在734 nm处测定。ABTS阳离子自由基清除活性的计算如公式(9)所示:
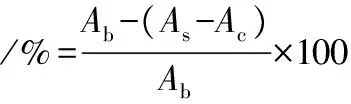
(9)
式中:As、Ac和Ab,样品、对照和空白的吸光度。
1.2.5.4 MTT法检测PC12细胞存活率
将PC12细胞密度为1×105个/mL,将其直接注射于96孔细胞培养板中,经过24 h的预培育后,将培养基吸去,加入100 mL无血清培养液,其中含有25 mmol/L PMSF,并设置对照组和空白组,每组4个重复实验。在PMSF处理24 h后,每一个孔中加入10 mL MTT溶解液,培育4 h后,然后将上清液吸出,向每一个孔内添加100 mL Formazan溶化液,混合均匀后放回细胞培养箱中孵育4 h以内,直到在倒置显微镜下观测不到紫色结晶为止。通过酶标仪检测570 nm下各孔的吸光度,并将其相应的存活率公式表示出来。存活率的计算如公式(10)所示:
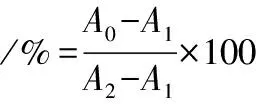
(10)
式中:A0、A1和A2,实验组、空白组和对照组OD值。
1.2.5.5 细胞内ROS含量测定
将PC12细胞以每孔1×105个/mL的密度注射于无菌的全黑96孔板中,待细胞生长融合至80%左右时,加入100 mL含有0、5、10、15、20、25 mmol/L PMSF的无血清培养液,经过24 h的PMSF处理后,取出旧的培育液,依次加入100 mL含量为10 mmol/L荧光探测器,以观察细菌的生长情况,并进行进一步的研究。在培养箱中孵育20 min,孵育完成后,利用PBS缓冲液进行1~2次冲洗,以除去未加入细菌的探针。最后,每个孔中加入100 mL PBS缓冲液,在阴暗的自然环境下,荧光强度用荧光酶标仪测定,激发波长和发射波长分别为495 nm和529 nm。经PMSF处理后,PC12细胞内ROS水平测定结果的计算如公式(11)所示:
(11)
式中:A0、A1和A2,实验组、空白组和对照组OD值。
1.2.5.6 细胞内抗氧化活性(antioxidant activity in the cell, CAA)测定
从PC12细胞中取出对数生长期的细胞,将其密度调节至6×104个/mL,然后将其均匀地分散于无菌的全黑96孔板中,在细菌培育完成后,应当除去孔内旧栽培基,并采用无菌D-Hank’s水溶液进行1~2次冲洗,以确保未充分贴壁和死去的细菌得到有效清除。为了进行比较,实验组中加入100 mL和DCFH-DA探针,对照组和空白组中分别加入100 mL稀释的DCFH-DA探针,以便进行更深入的研究。将细菌放入37 ℃、5% CO2栽培箱中孵育1 h,然后用无菌D-Hank’s水溶液冲洗2~3次,将试验组和对照组分别加入100 mL ABAP水溶液(600 mmol/L)和100 mL无菌D-Hank’s水溶液,接着迅速将其放入荧光酶标仪,设置5 min的间隔,实时检测1 h,以获取真实荧光信息。激发波长为485 nm,而发射波长为538 nm。CAA值的计算如公式(12)所示:
(12)
式中:SA,添加PMSF后的荧光值与时间形成曲线的面积;CA,空白组的荧光值与时间形成曲线的面积。
1.2.6 抑制癌细胞增殖
1.2.6.1 MTT法检测细胞活力
当细胞融合度达到90%时,采用传代操作方法获得细胞悬液。离心吸弃培养基,加入4 mL完全培养基后,用台盼蓝进行细胞染色,染色后对细胞计数,细胞均匀接种于96孔板中,密度为5×104个/mL,体积为90 mL。然后将96孔板在37 ℃的5% CO2培养箱中孵育一夜。将96孔板置于37 ℃ 5% CO2培养箱中48 h。给药培养48 h后,将MTT溶液依次加入96孔板,每孔体积为10 mL,将96孔板置于培养箱中继续培养4 h。每孔依次DMSO 100 mL。检测96孔板在570 nm和630 nm处的吸光度值。
1.2.6.2 细胞周期检测
用培养基配制不同浓度的cattle(125~2 000 mg/mL),每孔加入300 mL,将6孔板置于5 pc CO2培养箱中,37 ℃培养48 h。给药培养48 h后,先用200 mL不含EDTA的胰酶消化细胞,用离心机以2 000 r/min离心5 min收集细胞,吸弃上层液体,用预冷1×PBS进行洗涤,次数为2次,2 000 r/min离心5 min。弃上清液后,加入1 mL 75%乙醇,放入4 ℃冰箱过夜固定细胞。固定24 h后用离心机以2 000 r/min离心5 min收集细胞,吸弃上层液体,用预冷1×PBS进行洗涤,次数为2次,2 000 r/min离心5 min。再分别加入细胞周期检测试剂,在室温条件下避光孵育30 min,最后轻轻吹打均匀后再用流式细胞仪分析。
1.2.7 分子动力学
BCL-2、CDK-2蛋白晶体结构从Alphafold[21]数据库(https://alphafold.com/entry/A0A7L2ZRC1)中下载获得。小分子新橙皮苷结构从PubChem[22]数据库下载获得,并在MMFF94力场下进行能量最小化。分子动力学模拟采用AMBER 18软件进行模拟。模拟时,非键的截断距离设为10 Å,Particle mesh Ewald(PME)方法被用于计算长程的静电作用,SHAKE方法用于限制氢原子键长,Langevin算法用于温控,其中碰撞频率γ设为2 ps-1。体系压强为1 atm,积分步长为2 fs,每隔10 ps保存轨迹用于后续分析。
所有体系的蛋白和配体间的结合自由能通过MM/GBSA[23]方法计算。本研究中采用90~100 ns的MD轨迹用作计算,具体如公式(13)所示:
ΔGbind=ΔGcomplex-(ΔGreceptor+ΔGligand)
=ΔEinternal+ΔEVDW+ΔEelec+ΔGGB+ΔGSA
(13)
式中:ΔEinternal表示内能、ΔEVDW表示范德华作用、ΔEelec表示静电相互作用。其中内能包括键能(Ebond)、角能(Eangle)、和扭转能(Etorsion);ΔGGB和ΔGSA统称溶剂化自由能。其中,GGB为极性溶剂化自由能、GSA为非极性溶剂化自由能。对于ΔGGB,本文采用NGUYEN等[24]研究者开发得GB模型进行计算(igb=8)。非极性溶剂化自由能(ΔGSA)则基于表面张力(γ)与溶剂可及性表面积(surface area, SA)的乘积计算得到,ΔGSA=0.007 2×SA。熵变由于高耗计算资源与低精度,在本研究中忽略不计。
1.3 组分分析
基于液-质联用(liquid chromatograph mass spectrometer, LC-MS)非靶向的方式进行检测,重复3次,所得到的数据供生物信息学分析,最终得出代谢物。采用Thermo超高效液相系统(Vanquish,USA),根据化合物的性质,采用Hypersil GOLDTMC18选择性HPLC色谱柱,进样量10 mL,流速0.9 mL/min,柱温40 ℃,流动相A为85%甲酸-水(体积分数)溶液,流动相B为去离子水溶液。质谱采用的是美国Thermo公司的Qexactive高分辨质谱检测系统。
1.4 数据处理
实验中用Origin 2018来统计数据及绘图。
2 结果与分析
2.1 大孔树脂的筛选
2.1.1 吸附率与解吸率
吸附/解吸能力是评价回收目标化合物的吸附/解吸过程时最重要的指标。如图1所示,本实验共测试了6种大孔树脂,包括2种弱极性(AB-8和X-5),2种中极性(DM301和CAD-40)和2种非极性(D101和NKA)对PMSF的吸附/解吸能力。从图1可以看出,弱极性(AB-8和X-5)树脂对PMSF的吸附/解吸能力优于其他树脂,非极性(D101和NKA)树脂对PMSF的吸附/解吸能力相对较低。大孔吸附树脂的分离特性在很大程度上取决于其特定面积、表面极性和孔径[25],AB-8为聚苯乙烯型弱极性吸附树脂,表面有一定的酯基,亲水性得到改善,该树脂的比表面积和孔径较大,对于PMSF具有较好的吸附/解吸能力。
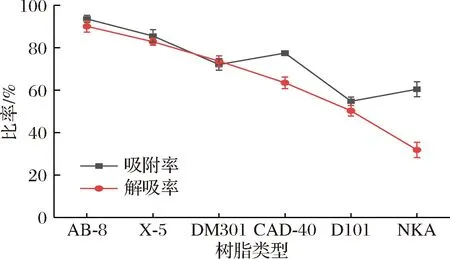
图1 六种大孔树脂吸附率与解吸率Fig.1 Adsorption and desorption rates of 6 macroporous resins
2.1.2 吸附动力学
吸附是一种物理化学过程[26],主要涉及溶质(吸附物)从液相到吸附剂表面的传质。树脂吸附[15]可以分为3个不同的区域:1)外部区域几乎完全饱和的吸收;2)中间区域在特定时间内发生的吸收;3)最内层区域尚未发生吸收。随着吸附过程的持续,饱和区越来越厚,从而促使颗粒内部的扩散过程。不同时间下的吸附动力学曲线如图2所示,AB-8大孔树脂的吸附性能优于其他5种。随着时间的增加,吸附量也逐渐提高。0~60 min时,PMSF吸附量迅速增加,此后,即使时间增加,吸附也不再改变,这是因为大孔树脂的表面结合位点大多已被总黄酮饱和。6种树脂的颗粒内扩散模型的线性回归如图3所示,AB-8树脂的颗粒内扩散模型(斜率)分别为0.003 95和0.075 71(表3)。很明显,AB-8树脂在吸附的初始阶段,颗粒内扩散并不是唯一的机制,因为回归线不是通过原点的。AB-8树脂的截距值为正,表明它具有快速动态的特性,因此,在初始阶段,吸附速率受到薄膜扩散等因素的影响,但粒内扩散仍然是主要机制[27]。
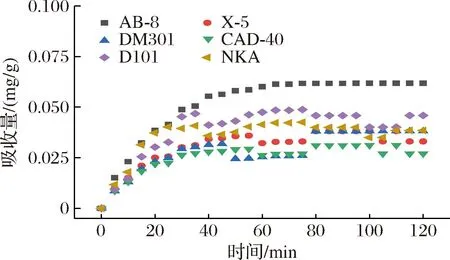
图2 六种大孔树脂吸附曲线Fig.2 Adsorption curves of 6 macroporous resins
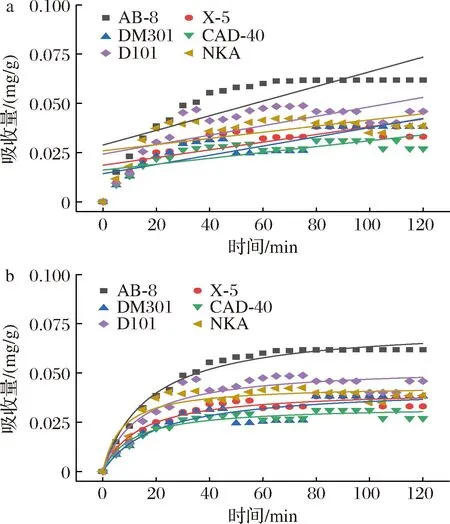
a-一级动力学方程;b-二级动力学方程
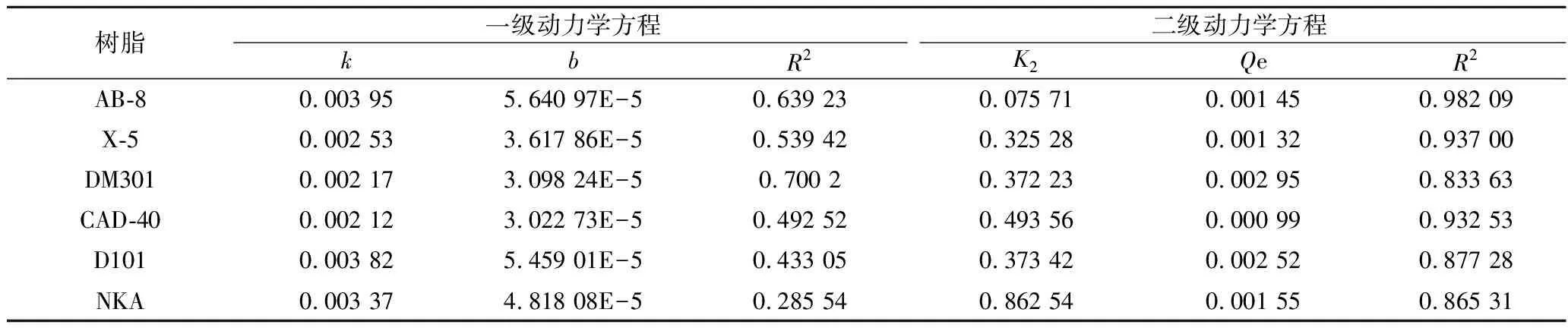
表3 一级动力学与二级动力学方程Table 3 First-order dynamics and Second-order dynamics equations
通过对6种树脂的吸附动力学方程拟合及Langmuir和Freundlich模型的分析,结果如图4所示,6种树脂对PMSF吸附能力最好的为AB-8树脂。Langmuir和Freundlich模型结果表明,AB-8树脂的吸附均适用于两种模型,Langmuir模型的R2为0.982 09,高于Freundlich模型的0.914 49(表4),该模型表明树脂表面是均质的。综合考虑,选用AB-8分离PMSF。

a-Langmuir等温吸附公式;b-Freundlich等温吸附公式
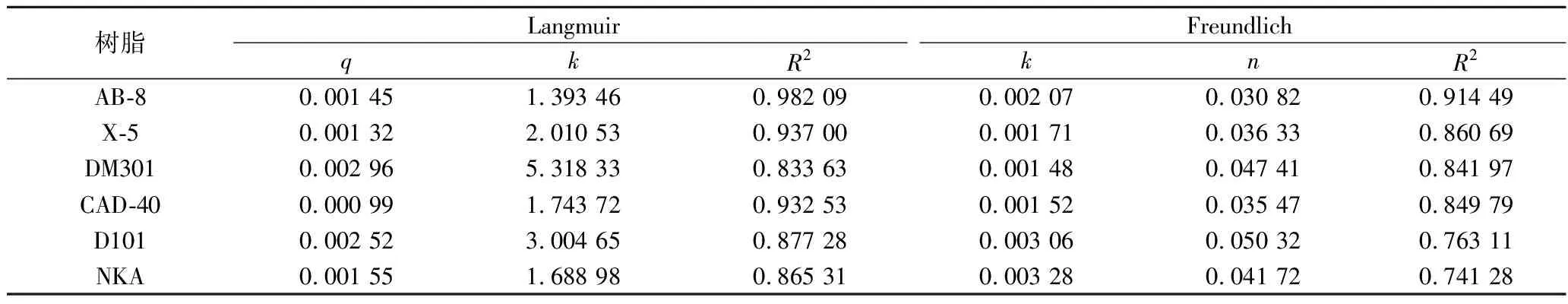
表4 Langmuir与Freundlich等温吸附公式Table 4 Langmuir and Freundlich isothermal adsorption formulas
2.2 动态洗脱工艺
上样速度、上样量的差异、洗脱剂的浓度以及洗脱流速的差异都会影响目标产物的最终分离效果。根据图5-a可以看出,上样流速对分离效果有显著影响。当上样流速为8 BV/h时,曲线迅速达到最高点,表明大孔树脂吸附已经达到饱和状态,PMSF未能被充分吸附进树脂内部就被冲出柱外。根据图5-b所示,当柱体积保持不变时,随着流出液的增加,PMSF的含量也会增加。当收集管数为60时,PMSF的含量达到最大值,为0.52 mg/mL,此时AB-8型大孔树脂的吸附已经接近饱和状态。由于改变乙醇溶液的浓度,可以有效地改变PMSF与大孔树脂溶液之间的疏水作用,进而使吸收物洗脱下来。根据图5-c所示,60%乙醇洗脱的PMSF含量最高达0.93 mg/mL,这可能是由于不同浓度的乙醇溶液对黄酮与大孔树脂间的作用力影响有所不同,因而使得洗脱效果更加显著。洗脱速度过快会导致分子无法从色谱柱中洗脱出来,太慢的洗脱速度则会导致多组分被一同洗脱出柱,分离效果受到影响,因此选择合适的洗脱速度也尤为重要。图5-d显示,当洗脱速度为5 mL/min时,PMSF含量最高达0.95 mg/mL。
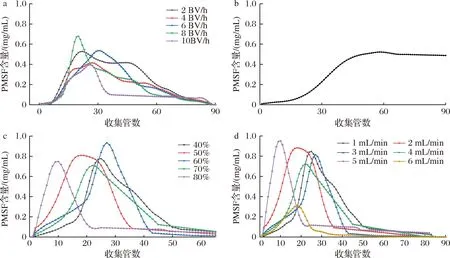
a-上样速度;b-上样量;c-洗脱剂;d-洗脱流速
2.3 C18分离
通过前期对于PMSF的分离,结合液相分析(图6-a),可以发现,PMSF有11个不同组分,AB-8树脂吸附后的液相图(图6-b)有3个不同组分,C18柱可有效分离新橙皮苷(图6-c),结合质谱鉴定(鉴定由苏州帕诺米克生物公司完成)(图6-d),新橙皮苷的质核比为610.18,分子结构为C28H34O15(图6-e),新橙皮苷的含量由PMSF中的27.21%提升至82.31%。

a-PMSF;b-AB-8组分;c-C18组分;d-新橙皮苷质谱;e-新橙皮苷结构
2.4 抗氧化活性
类黄酮是天然植物化学物质,可能具有治疗作用,有助于预防多种疾病。本实验测试了新橙皮苷(200、400、600、800、1 000 mg/mL)清除自由基能力(DPPH自由基、超氧阴离子自由基和ABTS阳离子自由基),结果如图7-a所示。
随着浓度的提高,新橙皮苷的抗氧化能力也逐渐提升,1 mg/mL DPPH、超氧阴离子和ABTS阳离子清除能力分别为83.23%、81.05%和72.03%。进行PC12活性测试,如图7-b所示,250 mg/mL添加量时,PC12细胞存活率高于85%,表明新橙皮苷对于PC12的活性影响不大,可进行细胞内抗氧化测试。通过检测添加了不同含量新橙皮苷的PC12细胞内活性氧(reactive oxygen species,ROS)含量,发现ROS含量随着新橙皮苷的添加而减少,当新橙皮苷添加量为250 mg/mL时,ROS含量较未添加减少了30.21%,CAA活性提高了14.39%,这表明新橙皮苷可以通过提高CAA活性来降低细胞内ROS含量。新橙皮苷的酚羟基结构是影响其抗氧化活性的关键因素,羟基基团的存在可以改善整体结合[28],并且可以促进分子与双层脂质膜的极性头部区域之间的相互作用;另一方面,酚羟基的存在可以抑制膜的疏水核心的转变,新橙皮苷的多个酚羟基基团,在细胞膜上建立脂水界面,可以阻止自由基进一步渗透到脂质疏水核心,从而来提高其细胞内抗氧化的能力。
2.5 抑制癌细胞增殖
本研究测试了质量浓度125~2 000 mg/mL下的新橙皮苷对于4种肿瘤细胞株(SH-SY5Y、K562、293T和MCF-7/ADR)的抑制能力,并用MTT法进行检测。如图8所示,2 000 mg/mL的新橙皮苷均可有效抑制4种肿瘤细胞株(SH-SY5Y、K562、293T和MCF-7/ADR),其中新橙皮苷对于K562的抑制能力最强,有效抑制率为92.49%,因此对K562进行MTT法检测,结果表明500 mg/mL时的新橙皮苷可以有效抑制K562的G1分裂时期从而实现抑制K562癌细胞增殖。
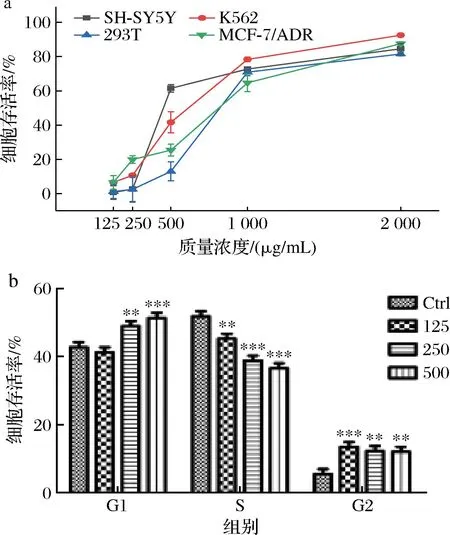
a-抑制4种癌细胞增殖能力;b-MTT法检测K562增殖
2.6 分子动力学
如图9-a所示,基于分子对接获取的小分子新橙皮苷与BCL-2蛋白的结合复合物,新橙皮苷结合在BCL-2蛋白的表面空腔内(见图9-a中1),从图9-a中2~3相互细节图中可以看出,结合位点由LEU-201、TYR-202、LEU-209、SER-216、ASN-143、PHE212氨基酸围绕而成的活性口袋。其中,小分子和位点上的LEU-201、LEU-209、PHE212发生疏水作用,和TYR-202、LEU-209、SER-216发生氢键作用。这些相互作用是小分子和蛋白稳定结合的主要原因。如图9-b所示,基于分对接获取的小分子新橙皮苷与CDK-2蛋白的结合复合物,新橙皮苷结合在CDK-2蛋白的表面空腔内(见图9-a中1),从图9-a中2~3相互细节图中可以看出,结合位点由PHE-87、PHE-85、ALA-34、ILE-88、VAL-13、VAL-21、GLY-16、LYS-93、THR-17、GLN-136、GLU-15、ASP-89、ASP-150、ASN-137氨基酸围绕而成的活性口袋。其中,小分子和位点上的PHE-87、PHE-85、ALA-34、ILE-88、VAL-13、VAL-21发生疏水作用,和ASP-150、ASN-137发生氢键作用。这些相互作用是小分子和蛋白稳定结合的主要原因。
分子动力学模拟的均方根偏差可以反应复合物的运动过程,RMSD越大以及波动越剧烈表示运动剧烈,反之,运动平稳。如图10所示,BCL-2、CDK-2结合了新橙皮苷的蛋白(红色)表现出比未结合新橙皮苷的BCL-2、CDK-2蛋白更低的RMSD。表明小分子与蛋白结合的非常密切,小分子与结合稳定性非常好。
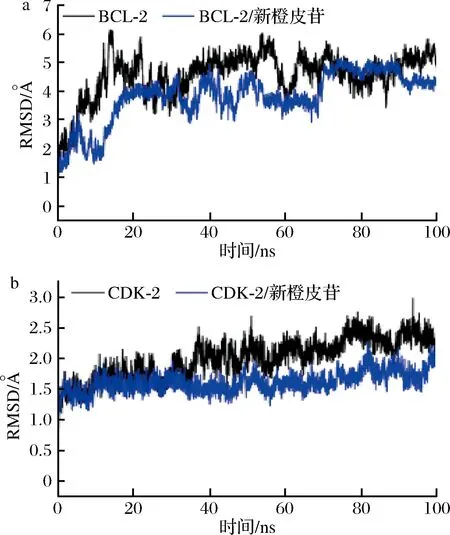
a-BCL-2;b-CDK-2
RMSF可以反应分子动力学模拟的过程中蛋白的柔性。通常药物结合蛋白后,蛋白柔性下降,进而达到稳定蛋白的作用,而发挥酶活性。如图11所示,新橙皮苷可以稳定BCL-2、CDK-2蛋白的整体柔性,进而达到潜在的影响BCL-2、CDK-2蛋白生物功能的效果。
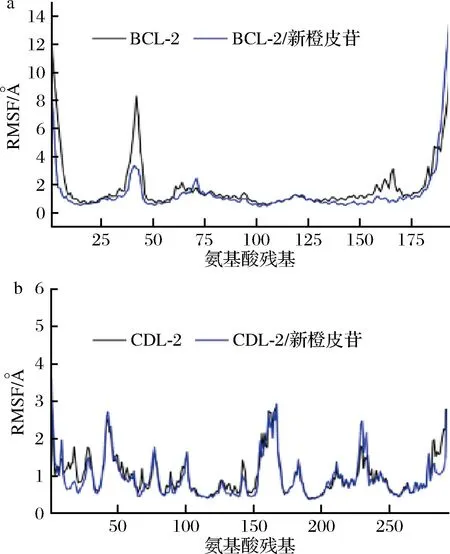
a-BCL-2;b-CDK-2
基于分子动力学模拟的轨迹,采用MM-GBSA的方法计算了结合能,该结合能可以更为准确的反应小分子和目标蛋白的结合效果。如表5所示,BCL-2/新橙皮苷、CDK-2/新橙皮苷的结合能为(-20.40±2.51) kcal/mol、(-44.18±1.77) kcal/mol。数值为负数表明该分子与目标蛋白具结合亲和力,数值越低表示结合越强。显然计算表明BCL-2/新橙皮苷、CDK-2/新橙皮苷的结合亲和力较强。通过能量分解,可以看出这些复合物的结合主要贡献能为静电能和范德华能,再就是非极性溶剂化自由能。

表5 MM/GBSA预测的结合自由能和能量成分 单位:kcal/mol
氢键为最强的非共价结合作用之一,数目越多表示结合越好。本研究监测了在模拟期间BCL-2/新橙皮苷、CDK-2/新橙皮苷复合物的氢键数目,以反应氢键在两者结合的贡献。如图12所示,在BCL-2/新橙皮苷复合物中,小分子和蛋白在模拟过程中形成的氢键数目在0~7个。在CDK-2/新橙皮苷复合物中,小分子和蛋白在模拟过程中形成的氢键数目在2~8个。大多数时间氢键数目在5~6,意味着新橙皮苷与CDK-2结合期间,氢键持续存在,并且时常发生多个氢键,对两者的结合起到重要贡献作用。相比较而言,新橙皮苷与CDK-2的氢键数目要大于与BCL-2蛋白的氢键数目,这和结合能效果一致。
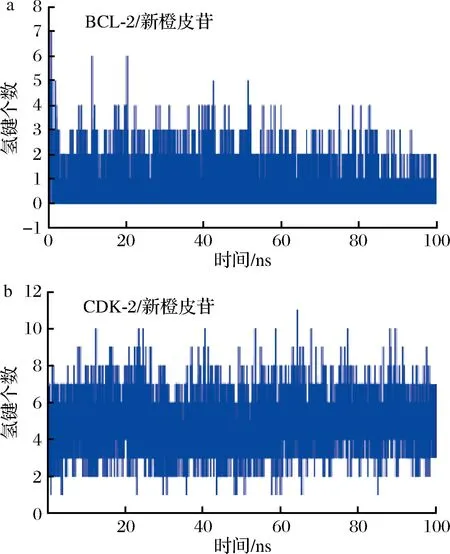
a-BCL-2;b-CDK-2
3 结论
牡丹籽粕作为牡丹籽榨油后的废弃物,存在很多活性物质(黄酮、多酚和蛋白等),本实验借助AB-8树脂联用C18柱的方法从牡丹籽粕中分离纯化出新橙皮苷,测试了新橙皮苷的生物活性,表明其具有优异的生物活性,1 mg/mL新橙皮苷对于DPPH自由基、超氧阴离子自由基和ABTS阳离子自由基清除能力分别为83.23%、81.05%和72.03%;细胞内抗氧化活性测试表明250 mg/mL新橙皮苷可以有效提高14.39% CAA活性,从而减少细胞内30.21% ROS含量。细胞内过量的ROS会促进癌细胞增殖,因其优异的抗氧化活性,因此测试了新橙皮苷对于抑制4种癌细胞(SH-SY5Y、K562、293T和MCF-7/ADR)增殖,实验表明2 000 mg/mL新橙皮苷对于K562的抑制率为92.49%,MTT法检测结果表明500 mg/mL新橙皮苷可以有效抑制K562的G1期;分子动力学模拟结果表明新橙皮苷可以通过结合BCL-2、CDK-2产生氢键抑制K562增殖。本实验从牡丹籽粕中分离纯化新橙皮苷,探究了新橙皮苷的生物活性,结果表明新橙皮苷具有良好的抗氧化活性与抑制K562癌细胞增殖能力,为科学认识牡丹籽粕废弃物提供理论基础。
猜你喜欢
阅读(快乐英语高年级)(2021年11期)2021-03-08
陶瓷学报(2020年6期)2021-01-26
制造技术与机床(2018年10期)2018-10-13
益寿宝典(2017年2期)2017-02-26
材料科学与工程学报(2016年2期)2017-01-15
吉林大学学报(医学版)(2015年3期)2015-12-17
汽车零部件(2015年1期)2015-12-05
中国当代医药(2015年24期)2015-03-01
中国当代医药(2015年9期)2015-03-01
中国氯碱(2014年12期)2014-02-28
