
儿童1p与16q共缺失相关性肾母细胞瘤1例
2024-05-12 10:52:18赵曼丽顾伟忠汤宏峰
临床与实验病理学杂志 2024年4期
刘 蕾,江 畅,赵曼丽,顾伟忠,汤宏峰
患者女性,2岁6个月,2021年11月检查发现右侧腹部肿块就诊。B超示:右后腹膜探及一低回声包块,大小14.8 cm×11.8 cm×11.7 cm,边界清,形态不规则,内回声不均匀,内见少量细小钙化斑及散在细小无回声区,与右肾关系密切,包块左下切缘可见残余肾组织,包块上缘紧贴肝右叶,与肝脏分界尚清,肝脏受压向左移位。包块后缘紧贴脊柱,左侧缘紧贴腹腔干及肠系膜上动脉,血管受压向左移位。包块边缘可见多枚低回声结节,皮髓质分界不清。下腔静脉局部受压变窄。CDFI示:低回声包块内可见较丰富血流信号。腹部增强CT示:右腹部可见较大混杂密度软组织块影,部分平扫肿块边界可辨,大小129.9 mm×113.6 mm×150.4 mm,平扫CT值为16~48 HU,肿块下方可见部分残余右肾结构,右肾形成抱球征,肿块包绕部分右肾动脉,可见右肾动脉分支及腹腔干分支进入肿块内部供血,形成多发强化小血管影,右肝动脉见分支走行于肿块边缘区。肿块推挤周围组织器官,肝脏、胰腺明显受推挤变形,胆囊位于左侧上中腹区,下腔静脉受推挤,向左前方移位,周围肠管受推挤左移。考虑右肾来源肾母细胞瘤,伴食管下段旁、腹膜后淋巴结转移,肝脏受累待排,左肾、输尿管、膀胱未见明显异常。相关辅助检查:LDH 4 699 U/L(正常范围:110~295 U/L),NSE 230.80 ng/mL(正常范围:0~23.26 ng/mL)。入院化疗后行右腹部肿瘤根治切除术及双侧疝囊高位结扎术,术后继续化疗。
病理检查眼观:右肾肿瘤根治性切除标本,肾切面见一肿物,总体积7.0 cm×5.5 cm×5.5 cm。镜检:大部分肿瘤细胞呈圆形或短梭形,部分呈巢状分布,核深染,核分裂象较多见(图1、2)。部分肿瘤细胞呈间叶型,肿瘤间可见软骨组织,未见间变,肿瘤部分坏死。肿瘤未侵及肾盂,区域肿瘤侵出包膜。骨髓常规:淋巴系统明显增生。免疫表型:CK(AE1/AE3)、vimentin、WT1(+),Cyclin D1、desmin(部分+),INI1、BRG1(存在),Pan-Trk(±),Bcor、S-100(-),Ki67(热点区域90%+)。FISH检测1q21和1p36基因探针显示:1p36红色信号缺失(缺失信号数/计数细胞数:130/200,65%)(图3);16q22/16q23基因缺失阳性(缺失信号数/计数细胞数:110/200,55%)(图4、5);WT1(11p13)基因缺失阴性(图6);SS18(SYT)基因断裂阴性;CIC(19q13)基因断裂阴性;EWSR1(22q12)基因断裂阴性。
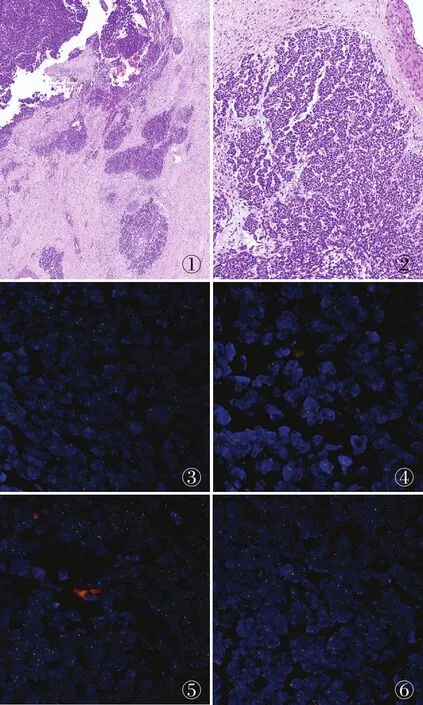
图1 肿瘤细胞呈巢状分布 图2 肿瘤细胞呈圆形或短梭形,核深染,核分裂象较多见 图3 FISH检测1q21和1p36基因探针(红色信号标记为1p36,绿色信号标记为1q21)显示1p36红色信号缺失 图4 FISH检测16q22基因探针(红色信号标记为16q22,绿色信号标记为着丝粒)显示缺失阳性 图5 FISH检测16q23基因探针(红色信号标记为16q23,绿色信号标记为着丝粒)显示缺失阳性 图6 FISH检测WT1(11p13)基因探针(红色信号标记为WT1,绿色信号标记为着丝粒)显示缺失阴性
病理诊断:恶性肿瘤,首先考虑肾母细胞瘤(Wilms’ tumor, WT)(胚芽为主型,非间变)。
讨论WT最常表现为腹部肿块,通常由父母发现,发病高峰年龄1~3岁,年发病率为1/10 000[1-2],术后长期生存率约为90%,部分患儿伴复发、转移或耐药等[3-4]。WT显微镜下具有三种不同发育阶段的肾脏组织学结构,包括间叶、上皮和原始胚芽组织三种成分。三种成分的比例及分化程度变化不一,因此,WT的组织学千变万化。有些肿瘤以原始胚芽为主,幼稚胚芽细胞为小圆形或卵圆形原始细胞,胞质少,似未分化小细胞肿瘤,其他两种成分可能很少。有些肿瘤以上皮组织为主,肿瘤有腺管样结构。也有些肿瘤以间叶成分为主,以不同分化程度的梭形细胞、横纹肌母细胞到成熟的横纹肌细胞为主。有时肿瘤中可混有少量成熟的鳞状上皮、软骨、骨或脂肪等组织。1p伴16q基因缺失(loss of heterozygosity, LOH)较罕见,由Maw等[5]于1992年首次报道,随后美国肾母细胞瘤协作组(National Wilms Tumor Study, NWTS)的NWTS-3和NWTS-4两项研究结果表明其与不良预后相关,NWTS-5研究结果表明1p伴16q LOH同时发生的情况占纳入总例数的5%,而英国Grundy等[6]研究占比则为2%。
本例大部分肿瘤细胞呈圆形或短梭形,巢状分布,部分肿瘤细胞呈间叶型,可见软骨组织,未见间变。结合免疫组化CK(AE1/AE3)、vimentin、WT1(+),Cyclin D1、desmin(部分+),INI1、BRG1(存在),Pan-Trk(±),Bcor、S-100(-),Ki67(热点区域90%+),符合胚芽为主型WT病理学特点。FISH检测1q21/1p36基因探针显示1p36红色信号缺失;16q22/16q23基因缺失阳性;WT1(11p13)基因缺失阴性,故本例为WT伴1p和16q共缺失。
染色体1p和16q杂合性缺失与WT患者的不良预后有关[7-8]。美国国家肾母细胞瘤研究NWTS-5(NCT00002611)发现染色体1p和16q共缺失WT患者的无事件生存率(event-free survival, EFS)和总生存率较低。参与研究的1 727例组织型良好的肾母细胞瘤(favorable histology Wilms’ tumor, FHWT)中,11%为1p缺失,17%为16q缺失,5%为1p和16q共缺失。LOH组的4年EFS为74.9%,其中LOH为1p和16q共缺失患者为65.9%,无缺失组为91.2%[9]。Ⅰ期(完全切除的局限性疾病)FHWT患者,年龄<24个月、肾切除重量<550 g的患者单独进行手术切除;年龄≥24个月或肾切除重量>550 g的Ⅰ期FHWT患者和Ⅱ期(全切除并有区域内传播)患者接受EE4A方案治疗,其中包括给予长春新碱和放线菌素D超过22周的治疗;Ⅲ期(肿瘤局限于腹部)或Ⅳ期(腹部外转移)FHWT患者采用DD4A方案治疗,包括添加阿霉素和长春新碱和放线菌素D,疗程超过28周[10]。只有LOH为1p合并16q时,Ⅲ期或Ⅳ期患者的复发和死亡风险才会增加。在整个研究中,LOH 1p合并16q的患者对结果的影响最大[6]。
WT除间变型和非间变型需进行鉴别,还需与其他肾脏肿瘤相鉴别,如神经母细胞瘤和淋巴瘤等小细胞肿瘤、肾横纹肌样肉瘤、肾源性残余等。WT具有较广的组织学形态谱,可以表现类似上述疾病的病理形态,因此需借助免疫组化和分子病理等方法鉴别。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:22
现代临床医学(2021年6期)2021-11-20 06:34:38
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:54
家庭医学(下半月)(2019年11期)2020-01-16 08:39:06
中国生殖健康(2019年9期)2019-01-07 01:19:00
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
海南医学(2016年8期)2016-06-08 05:43:00
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
物理实验(2015年9期)2015-02-28 17:36:47
中国医疗美容(2015年5期)2015-02-03 03:02:01
