
通过式固相萃取-超高效液相色谱-串联质谱法测定食品中的氯米芬、曲美他嗪和美度铵
2024-05-08 03:00:46张阳阳姚蕾珺肖全伟李绍波
食品科学 2024年8期
宋 娟,李 君,张阳阳,姚蕾珺,肖全伟,李绍波,*,戴 琴,*
(1.成都市食品检验研究院,四川 成都 611130;2.国家市场监管重点实验室(营养与健康化学计量及应用),北京 100029)
氯米芬是具有顺、反异构的N,N-二乙基-2-[4-(1,2-二苯基-2-氯乙烯基)苯氧基]乙胺化合物,稳定性较差,通常以枸橼酸盐形式存在[1-2]。盐酸曲美他嗪属哌嗪类衍生物,化学名为1-(2,3,4-三甲氧基苯甲基)哌嗪二盐酸盐,是一种抗心肌缺血的代谢性药物,具有降低血管阻力、增加冠脉及循环血流量、促进心肌代谢、降低心肌耗氧量、改善心肌氧的供需平衡、增强心脏功能的作用[3]。美度铵,又称米屈阱,最初作为一种生长促进剂用于动物养殖,此外,作为左旋肉碱抑制剂,在欧洲作为临床药物治疗糖尿病、神经退行性疾病和支气管肺疾病。就运动表现而言,对运动员运动能力、耐力以及运动后恢复均有促进作用[4]。因此,2016年,世界反兴奋剂中心将美度铵列入《禁用清单》S4激素及代谢调节剂。为了避免食品来源的兴奋剂污染,国家体育总局也于2021年发布《大型赛事食源性兴奋剂防控工作指南》,要求大型赛事举办方对食品中的食源性兴奋剂进行检测,把氯米氛、曲美他嗪和美度铵同时归为代谢调节剂。其中氯米芬的参考检出限为1 μg/kg;曲美他嗪的参考检出限为2 μg/kg;美度铵的参考检出限为20 μg/kg。检测对象为畜禽肉及其制品、水产品及其制品、蛋及其制品、奶及其制品,除此之外,曲美他嗪的检测对象还包括所有经厂家加工过的烹调调味品(如油、盐、酱、醋、料酒、味精、鸡精、蚝油等),检测对象多,基质较为复杂。
现有氯米芬、曲美他嗪和美度铵的检测方法主要包括液相色谱法、液相色谱-串联质谱法、超临界色谱-串联质谱法、液相色谱-高分辨质谱法等[5-14]。液相色谱检测法分析时间长,且易受基质干扰,满足不了痕量分析的需要;超临界色谱-串联质谱法分析速度快,但正相色谱柱易受样品中水的影响,不适合长期、大批量样品的分析。质谱仪作为高效液相色谱的检测器,大大提高了仪器的灵敏度和检测范围。三重四极杆质谱仪具有高效的对已知目标化合物定量检测的能力,目前已广泛应用于食品中多种药物残留的检测。通过多反应离子监测的扫描模式,能大大减少复杂基质样品中的背景干扰,提高目标化合物的检测灵敏度,保证分析结果的准确性。
目前,氯米芬、曲美他嗪和美度铵的绝大多数检测方法是建立在药品、尿液及血液基质中,食品中的检测方法报道较少。由于食品样品种类复杂,基质干扰效应明显,因此对样品前处理方法提出了更高的要求。而目前还鲜有食品中同时检测氯米芬、曲美他嗪、美度铵3 种代谢调节剂的报道。
1 材料与方法
1.1 材料与试剂
枸橼酸氯米芬(纯度99.0%)、曲美他嗪(纯度99.9%)、美度铵(99.0%)、氯米芬-D5盐酸盐(纯度98.5%)、曲美他嗪二盐酸盐-D8(纯度98.7%)和美度铵-D3(100.0 μg/mL)的标准品购于天津阿尔塔科技有限公司。
甲酸、甲醇、乙腈、甲酸铵、正己烷(均为色谱纯)德国Merck公司;氯化钠、二甲基亚砜(均为分析纯)重庆川东化工(集团)有限公司;Oasis PRiME HLB固相萃取小柱(200 mg/6 mL)美国Waters公司;安捷伦Elut净化包 美国Agilent公司;实验用水符合GB/T 6682—2008《分析实验室用水规格和试验方法》实验室一级用水。本研究过程中所用100余批次样品均购自于市场。
1.2 仪器与设备
1290+6470超高效液相色谱-三重四极杆串联质谱仪美国Agilent公司;Centrifuge 5810R高速离心机 德国Eppendorf公司;Milli-Q超纯水仪 美国Millipore公司;Multi reax全能型振荡器 德国Heidolph公司;BP211D型天平 德国Sartorius公司;ORTEX3旋涡混匀器 德国IKA公司。
1.3 方法
1.3.1 标准物质的配制
1.3.1.1 标准溶液配制
标准储备溶液:准确称取适量枸橼酸氯米芬、曲美他嗪和美度铵标准品,分别用甲醇溶解并定容至刻度,摇匀,配制成氯米芬(折算成枸橼酸后)、曲美他嗪和美度铵质量浓度各为1 000 μg/mL的标准储备液,-18 ℃保存。
混合标准中间溶液:分别准确吸取不同体积的氯米芬、曲美他嗪和美度铵标准储备溶液于同一棕色容量瓶中,用90%乙腈-水溶液(含0.1%甲酸)定容至刻度,摇匀得混合标准中间溶液(氯米芬、曲美他嗪质量浓度为0.2 μg/mL,美度铵质量浓度为1.0 μg/mL)。
1.3.1.2 同位素内标标准溶液配制
同位素内标储备液:分别准确称取氯米芬-D5盐酸盐、曲美他嗪二盐酸盐-D8和美度铵-D3标准品适量(准确至0.01 mg)置于10 mL容量瓶中,用甲醇溶解并定容配制成氯米芬-D5(折算盐酸后)、曲美他嗪-D8(折算盐酸后)和美度铵-D3质量浓度各为100 μg/mL的同位素内标储备液,-18 ℃保存。
混合同位素内标中间工作液:分别准确吸取不同体积的氯米芬-D5、曲美他嗪-D8和美度铵-D3同位素内标储备液至同一棕色容量瓶中,用90%乙腈-水溶液(含0.1%甲酸)定容至刻度,摇匀得混合同位素内标标准溶液(氯米芬-D5、曲美他嗪-D8质量浓度为0.2 μg/mL,美度铵-D3质量浓度为1.0 μg/mL)。
1.3.1.3 标准溶液配制
分别吸取不同体积的混合标准中间溶液和500 μL的混合同位素内标标准溶液于不同10 mL容量瓶中,用90%乙腈-水溶液(含0.1%甲酸)定容至刻度,摇匀,待测。
1.3.2 样品前处理
1.3.2.1 提取
固态和半固态样品:准确称取均质样品2 g(精确至0.01 g),置于50 mL离心管中,加入100 μL混合同位素内标中间工作液,加入均质子、5 mL水,涡旋混匀5 min。加入20 mL 5%甲酸-乙腈溶液,涡旋混匀10 min,超声30 min,再加入8 g氯化钠,涡旋混匀1 min,于9 500 r/min、4 ℃冷冻离心5 min,取上清液于另一离心管中,加入10 mL乙腈饱和正己烷溶液,涡旋混匀5 min,于9 500 r/min、4 ℃冷冻离心5 min,弃去正己烷层,下层溶液待净化。
液态样品:准确称取均质样品2 g(精确至0.01 g),置于50 mL离心管中,加入100 μL混合同位素内标中间工作液,加入3 mL水,涡旋混匀5 min。加入20 mL 5%甲酸-乙腈溶液,涡旋混匀10 min,超声30 min,再加入8 g氯化钠,涡旋混匀1 min,于9 500 r/min、4 ℃冷冻离心5 min,取上清液于另一离心管中,加入10 mL乙腈饱和正己烷溶液,涡旋混匀5 min,于9 500 r/min、4 ℃冷冻离心5 min,弃去正己烷层,下层溶液待净化。
1.3.2.2 净化
取10 mL下层溶液过PRiME HLB小柱,收集滤液于氮吹管中,加入50 μL二甲基亚砜,40 ℃氮吹至近干,用90%乙腈-水溶液(含0.1%甲酸)定容至1 mL,混匀,过0.22 μm有机滤膜,上机待测。
1.3.3 超高效液相色谱-串联质谱条件
1.3.3.1 超高效液相色谱条件
色谱柱:ACQUITY UPLC BEH Amide(3.0 mm×100 mm,1.7 μm);流动相A为0.1%甲酸-乙腈溶液,B为20 mmol/L甲酸铵溶液(含0.1%甲酸);流速:0.4 mL/min;柱温:40 ℃;进样量:2 μL;梯度洗脱程序见表1。
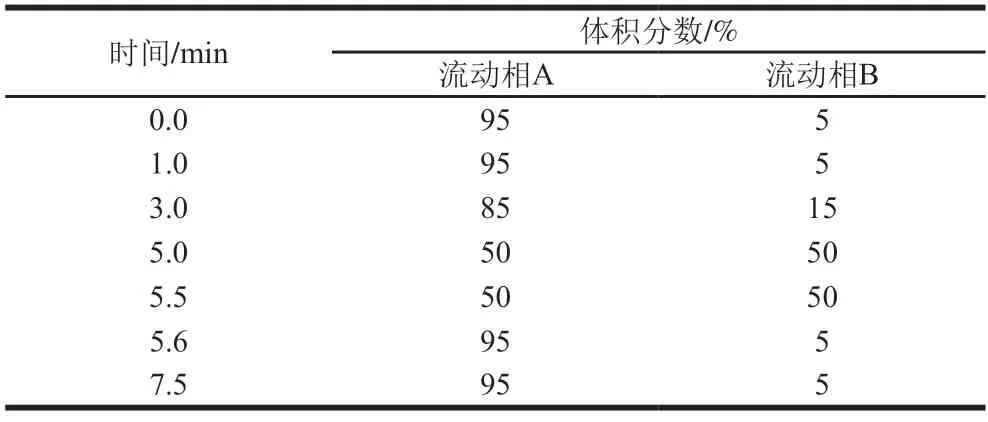
表1 梯度洗脱程序Table 1 Gradient elution program
1.3.3.2 质谱条件
离子源:电喷雾离子源(ESI+);检测方式:多重反应监测模式;干燥气(氮气)温度:300 ℃;干燥气(氮气)流量:10 L/min;雾化气(氮气)压力:35 psi;鞘气温度:300 ℃;鞘气(氮气)流量:11 L/min;毛细管电压:3 500 V;监测离子对和质谱参数见表2。

表2 3 种代谢调节剂检测的质谱条件Table 2 Mass spectrometric parameters for three metabolic regulators
1.4 数据处理
采用MultiQuant 3.0.2数据处理系统对样品中的3 种代谢调节剂进行超高效液相色谱-串联质谱分析、数据采集和处理,采用Origin 2023进行统计计算和图表绘制。
2 结果与分析
2.1 质谱条件优化
分别将100 μg/L单一标准溶液注入离子源,进行一级质谱全扫描,在最佳碎裂电压下确认目标化合物的母离子m/z值,然后进行二级质谱优化,优化碰撞能量,对碎片离子进行全扫描,选取响应最强与次强的碎片离子作为定量和定性离子[15]。
实验发现所有被测物均在ESI+下具有较强的信号值,3 种代谢调节剂均可获得较高丰度的[M+H]+峰。在优化干燥气温度、干燥气流量、雾化气压力、鞘气温度、毛细管电压等离子源条件后,得到质谱条件参数,结果见表2。
2.2 色谱条件优化
2.2.1 色谱柱选择
本研究考察了ACQUITY UPLC BEH C18(3.0 mm×100 mm,1.7 μm)、ACQUITY UPLC BEH HILIC(3.0 mm×100 mm,1.7 μm)和ACQUITY UPLC BEH Amide(3.0 mm×100 mm,1.7 μm)3 种色谱柱对待测化合物的分离效果。在同一流动相条件下,美度铵在C18色谱柱出峰速度过快,峰形较差,且易被内源性杂质(如乙酰胆碱同位素、赖氨酸和谷氨酸等)干扰[12];采用HILIC柱,美度铵出峰时间靠后,峰形较宽;而采用Amide柱,3 种目标物出峰时间、峰形和分离度均可以满足要求。因此,最终选择ACQUITY UPLC BEH Amide(3.0 mm×100 mm,1.7 μm)作为目标物分离的色谱柱,美度铵在C18柱、Amide柱和HILIC柱上的色谱峰比较见图1。

图1 美度铵在C18柱(A)、Amide柱(B)和HILIC柱(C)上的色谱峰比较Fig.1 Comparison of chromatographic peaks of meldonium on C18 (A),Amide (B) and HILIC (C) columns
2.2.2 流动相的选择
选择乙腈作为流动相的有机相,并加入0.1%甲酸优化峰形。考察5 mmol/L甲酸铵(含0.1%甲酸)、10 mmol/L甲酸铵(含0.1%甲酸)、20 mmol/L甲酸铵(含0.1%甲酸)、50 mmol/L甲酸铵(含0.1%甲酸)4 种不同浓度的甲酸铵溶液作为流动相对3 种物质的分离效果影响,结果表明4 种溶液均能达到目标物出峰、分离的要求,综合考虑峰形、对称度及流动相含盐量,本方法选择20 mmol/L甲酸铵(含0.1%甲酸)溶液作为流动相的水相。3 种化合物的总离子流色谱图见图2。

图2 3 种代谢调节剂标准溶液总离子流色谱图Fig.2 Total ion current chromatograms of mixed standard solution of three metabolic regulators
2.3 提取条件优化
目前针对该类物质的提取溶剂有甲醇、甲醇-水、乙腈、乙腈-水等。乙腈具有沉淀蛋白、与油脂相容性较差等特点[16],本研究选择乙腈作为基础提取溶剂,分别用乙腈、0.5%甲酸-乙腈、1%甲酸-乙腈、2%甲酸-乙腈、5%甲酸-乙腈、6%甲酸-乙腈对猪肉中3 种代谢调节剂的提取效率进行考察,结果见图3。结果表明,乙腈中加入甲酸可提高3 种目标物的提取效率,且随甲酸含量增加美度铵的提取效率明显提高,甲酸体积分数大于5%后,提取效率无明显变化。本研究选择5%甲酸-乙腈作为提取溶剂。
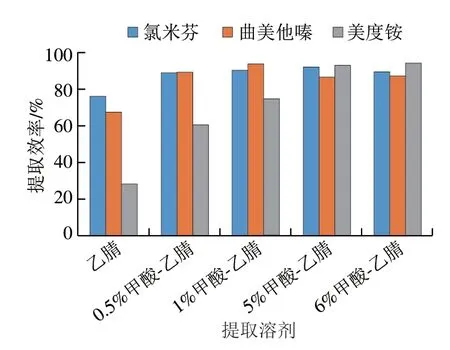
图3 5 种提取溶剂的提取效率对比Fig.3 Comparison of extraction efficiencies of five solvents
2.4 净化方式优化
由于食品中含有大量脂溶性杂质、色素等干扰物,容易干扰仪器检测,因此选择适宜的净化方式对方法的建立尤为关键。目前主流的净化方式有固相萃取小柱[17-18]、QuEChERS[19-21]、液液分散萃取[22]等净化方式。本研究分别考察了QuEChERS(C18100 mg+乙二胺-N-丙基硅烷(primary second amine,PSA)硅胶100 mg+石墨化碳50 mg)、安捷伦Elut净化包和PRiME HLB小柱(200 mg/6 mL)的净化效果,以猪肉为基质的净化效果见图4。结果表明,3 种净化方式对氯米芬的回收率影响差别不大。而采用QuEChERS和安捷伦Elut净化包两种净化方式时,曲美他嗪回收率大于120%;由于内源性的杂质干扰严重[12],采用QuEChERS方式净化时,美度铵回收率甚至大于200%。PRiME HLB小柱填料是一种新型的反相固相萃取吸附剂,具有免活化,操作简单,能有效去除蛋白、盐、磷脂等95%以上的基质干扰物,使样品基质效应更小等优点[23-25]。本研究在采用PRiME HLB小柱净化时,美度铵的内源性杂质干扰信号明显降低,3 种目标物的回收率在80%~110%之间,均满足GB/T 27404—2008《实验室质量控制规范》对回收率的要求。因此采用PRiME HLB小柱作为本研究的净化方式。
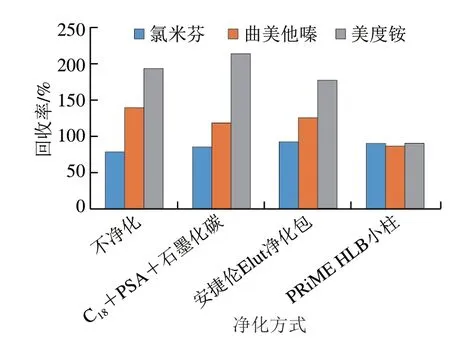
图4 不同净化方式的回收率对比Fig.4 Comparison of recoveries of different purification methods
2.5 基质效应
使用超高效液相色谱-串联质谱法检测食品过程中可能带入不同程度的基质效应,基质效应的存在直接影响到检测结果的准确性[26-29],因此本研究将对所建立方法的基质效应进行评估。
称取适量份数的17 种空白基质样品,除不加入同位素内标标准溶液外均按照1.3.2节获取空白样品提取液。分别使用空白样品提取液和90%乙腈-水溶液(含0.1%甲酸)按照1.3.1.3节配制标准曲线工作液,以基质标准曲线斜率与溶剂标准曲线斜率的比值考察3 种代谢调节剂的基质效应。若基质标准曲线斜率与溶剂标准曲线斜率的比值在0.85~1.15之间,则认为不存在基质效应的影响,反之则存在基质效应。3 种目标物的基质效应结果见图5。结果表明在17 种基质中3 种代谢调节剂均存在较大的基质效应,因此本研究采用同位素内标法做定量分析,从而减少基质效应对实际样品测定的干扰。
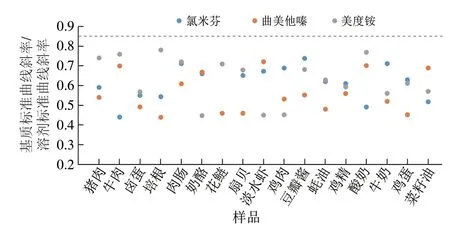
图5 基质效应考察Fig.5 Evaluation of matrix effect
2.6 线性关系、检出限与定量限
采取在空白试样中逐级降低加标浓度的方式确定3 种代谢调节剂的检出限和定量限。同时在不同基质中加入检出限和定量限水平的标准物质,按照1.3.2节和1.3.3节的方法进行检测。结果表明不同基质中检出限水平的各代谢调节剂其定性离子对、定量离子对信噪比均大于3;定量限水平的各代谢调节剂其定性离子对、定量离子对信噪比均大于10。氯米芬和曲美他嗪在1.0~40.0 ng/mL质量浓度范围内线性关系良好;美度铵在5.0~200.0 ng/mL质量浓度范围内线性关系良好。相关结果见表3。

表3 3 种被测物对应的内标化合物、线性范围、线性方程、决定系数、检出限及定量限Table 3 Internal standards,linear ranges,linear equations,determination coefficients (R2),limits of detection and limits of quantification of three metabolic regulators
2.7 方法回收率与精密度
依据GB/T 27404—2008《实验室质量控制规范食品理化检测》,分别称取猪肉、肉肠、牛肉、培根、鸡肉、牛奶、酸奶、奶酪、鸡蛋、卤蛋、花鲢、扇贝、淡水虾、豆瓣酱、蚝油、鸡精、菜籽油空白基质样品,按照定量限、2 倍定量限和10 倍定量限进行3水平添加实验(n=6),按照1.3.2节和1.3.3节方法进行样品处理和检测,考察所确定方法的回收率和精密度[30]。如表4所示,本方法的平均回收率在81.3%~100.7%之间,相对标准偏差(relative standard deviation,RSD)在0.3%~6.1%之间。

表4 方法加标回收率及RSD(n =6)Table 4 Recoveries and RSDs of the developed method (n=6)
2.8 实际样品测试
采用本方法对购自于市场的畜禽肉及含畜禽肉制品、水产品及含水产制品、蛋及含蛋制品、奶及含奶制品、植物油及含植物油制品以及烹调调味料等共100余批次样品进行3 种代谢调节剂的检测,均未检出阳性样品。
3 结论
本研究建立了通过式固相萃取-超高效液相色谱-串联质谱的检测方法,分别对前处理方法、色谱和质谱条件进行优化,采用多反应监测模式检测,同时测定食品中的氯米氛、曲美他嗪和美度铵。该方法具有灵敏度高、普适性强、操作简便等优点,可为体育赛事的食品保障工作提供有效的技术支撑。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
中国特种设备安全(2021年12期)2021-04-26 14:37:00
中成药(2018年6期)2018-07-11 03:01:32
中国蜂业(2018年4期)2018-05-09 06:25:08
现代园艺(2018年3期)2018-02-10 05:18:22
当代化工研究(2016年6期)2016-03-20 16:21:46
中国粮油学报(2016年5期)2016-01-23 02:45:06
中国继续医学教育(2015年5期)2016-01-07 07:38:27
无机化学学报(2014年3期)2014-02-28 17:30:58
河南科技(2014年12期)2014-02-27 14:10:32
