
Hepatocellular carcinoma immune microenvironment and check point inhibitors-current status
2024-05-08 09:21TaranaGuptaNikhilSaiJarpula
World Journal of Hepatology 2024年3期
Tarana Gupta, Nikhil Sai Jarpula
Abstract Hepatocellular carcinoma (HCC) is the most common primary tumor of the liver and has a high mortality rate. The Barcelona Clinic Liver Cancer staging system in addition to tumor staging also links the modality of treatment available to a particular stage. The recent description of the tumor microenvironment (TME) in HCC has provided a new concept of immunogenicity within the HCC. Virusrelated HCC has been shown to be more immunogenic with higher expression of cytotoxic T lymphocytes and decreased elements for immunosuppression such as regulatory T cells. This immunogenic milieu provides a better response to immunotherapy especially immune checkpoint inhibitors (ICIs). In addition, the recent data on combining locoregional therapies and other strategies may convert the less immunogenic state of the TME towards higher immunogenicity. Therefore, data are emerging on the use of combinations of locoregional therapy and ICIs in unresectable or advanced HCC and has shown better survival outcomes in this difficult population.
Key Words: Hepatocellular carcinoma; Tumor immune microenvironment; Immune checkpoint inhibitor; Atezolizumab; Bevacizumab; Pembrolizumab
INTRODUCTION
Primary as well as metastatic carcinoma can be found in the liver. Liver cancer is the sixth most common cancer worldwide and ranks fourth in the list of cancer-related deaths. It has a dismal 5-year survival of 18%[1]. Hepatitis B which is a carcinogenic virus has remained among the most common causes of hepatocellular carcinoma (HCC), especially in China. However, with universal immunization programs and hepatitis C elimination programs, alcohol and metabolic dysfunction associated steatotic liver diseases are emerging etiologies of HCC worldwide.
The Barcelona Clinic Liver Cancer (BCLC)[2] guidelines, first proposed in 1999, are the most accepted and practiced guidelines for prognostication and management of HCC. The recent BCLC 2022 update[3] has further clarified the grey areas in different stages of HCC, downstaging of tumors, treatment stage migration and progression of HCC within the same stage. “Untreatable progression” represents failure of the selected treatment strategy or progression of disease but remains in the same stage resulting in the need for consideration of therapy for a more advanced stage. This led to new staging upon progression after initial diagnosis, which includes 3 groups BCLCp-B defined as initially stage B and progressed but remained in stage B, BCLCp-C1 shows growth of the existing lesion or new lesions in the liver only. If there is new vascular invasion or new extrahepatic sites of metastases this is considered BCLCp-C2.
The pathophysiology of HCC is intricately linked to chronic liver diseases, which are characterized by prolonged hepatocytic injury and inflammation resulting in repair and regeneration of hepatocytes. These repeated cycles of injury and repair lead to genetic mutations such as in telomerase reverse transcriptase, catenin beta-1, tumor protein 53 (TP53), axis inhibition protein 1, AT-rich interaction domain 1A (ARID1A) and ARID2[4,5]. These mutations affect the cell cycle control and wingless-related integration site (WNT)-beta-catenin pathway which in addition to epigenetic mechanisms result in activation of hepatocarcinogenic pathways. Unlike other solid tumors, no single gene mutation is attributable to HCC development. Systemic therapies have been an integral part of the management of advanced HCC (BCLC stage C), especially tyrosine kinase inhibitors (TKIs) [Sorafenib 2007 and lenvatinib (LEN) since 2018]. They have improved outcome in HCC[6-8] and other TKIs such as regorafenib and cabozantinib are used in the second-line treatment of advanced HCC.
Tumor growth and regression also depend on interaction of the immune system with cancer cells, where cancer cells employ mechanisms to evade the immune system such as by downregulating the major histocompatibility complex (MHC) or expressing the immune checkpoint proteins like programmed death receptor ligand-1 (PD-L1) and programmed cell death protein-1 (PD-1). This has led to the development of therapies targeting these molecular and immune mechanisms.
TUMOR lMMUNE MlCROENVlRONMENT
HCC is almost a prototype of inflammation-associated cancer. The tumor microenvironment (TME) has both cellular and non-cellular components. The cellular component has damaged hepatocytes, hepatic progenitor cells and different types of immune cells. The non-cellular component has tumor stroma with growth factors, inhibitory factors, proteolytic enzymes and both pro-inflammatory and anti-inflammatory cytokines. The TME is also dependent upon and modulated by the etiology of chronic liver disease, genetics, epigenetics and other factors related to cellular metabolism.
The liver plays a pivotal role in immune regulation with its large reservoir of immunocompetent cells including neutrophils, monocytes, Kupffer cells, natural killer (NK) cells, dendritic cells (DCs) and lymphocytes (B lymphocytes, CD4+, CD8+). To maintain homeostasis, the liver environment always has a balance between pro-inflammatory [Interleukin (IL)-2, IL-7, IL-12, IL-15, and interferon γ (IFN-γ)] and anti-inflammatory mechanisms [IL-10, IL-13, and transforming growth factor β (TGF-β)][9]. In chronic liver diseases, there is an inclination towards pro-inflammatory signals due to necroinflammation in hepatocytes. Also, the abnormal gut-microbiota-liver axis increases the risk of bacterial infections in patients with cirrhosis leading to the production of anti-inflammatory cytokines such as IL-10 by Kupffer cells and DCs in the liver which suppress the co-stimulatory molecules on antigen presenting cells preventing CD4+T cells activation[10,11]. T cell mediated immunity is also decreased in chronic hepatitis B whereas hepatitis C evades the immune system of the host due to its high mutational rates and through viral factors that counteract DNA sensors[12,13]. Hence, the microenvironment in cirrhosis is a combination of inflammation and immunosuppression forming a safe niche for cancer cells to grow and counteract the immune mechanisms.
Immune activation
Due to tumor cell proliferation, necrosis and lately due to treatment, cancer cell antigens are released continuously. These antigens are captured by DCs through interaction with toll-like receptor (TLR)2 and TLR4. The DCs undergo maturation and under the influence of chemokines migrate to the lymph nodes[14,15]. Following the activation of co-stimulatory molecules CD40 on DCs, these antigens are presented to CD8+cytotoxic T lymphocytes (CTLs) in lymph nodes. Additionally, CTLs are also activated by IFN-γ released from NK cells, TH1 cells and tumor necrosis factor (TNF)-α and IL-12 released from macrophages and chemokines (CXCL-9, CXCL-10, CCL-5). With the interaction between lymphocyte function associated antigen 1 on activated T lymphocytes and intercellular adhesion molecule 1, tumor cells can be infiltrated by CTLs. Subsequently with recognition of cancer cells by T cell receptors along with co-stimulatory receptors, activated CTLs kill the cancer cells[16] (Figure 1).

Figure 1 lmmune activation in the tumor microenvironment. Interaction between tumor cells and the immune system is demonstrated in this figure. Antigen presentation by dendritic cells (antigen presenting cells) leads to activation of cytotoxic T lymphocytes which eventually leads to death of tumor cells by the release of granzymes and perforins. Activation of cytotoxic T cells require additional co-stimulatory signals during the interaction between dendritic cells and cytotoxic CD8+ T lymphocytes. PD-1: Programmed death receptor-1; PD-L1: Programmed death receptor ligand-1; ICI: Immune checkpoint inhibitor; TCR: T-cell receptor; MHC-1: Major histocompatibility complex I.
On the contrary, various check point molecules such as CTL-associated protein 4 and PD-1 bind to the CD80/86 molecule and interact with PD-L1 on DCs, respectively, and suppress the immune response. Immune-inhibiting cytokines such as IL-10, TGF-β, prostaglandin E2 (PGE2), and indoleamine 2,3-dioxygenase influence the expression of PD-1 on T cells and PD-L1 on DCs[17]. Additionally, vascular endothelial growth factor (VEGF) phosphatase and tensin homolog deleted on chromosome 10 produced by cancer cells activate the phosphotidylinositol 3/AKT pathway to suppress T cell infiltration[18].
Immune suppression
Tumor associated neutrophils:Neutrophils are a vital component of the immune system playing important roles during infection, injury and tumorigenesis. In the TME, tumor associated neutrophils (TANs) are recruited through the release of CXCL-5 and CXCL-6[19]. These neutrophils have a key role in tumor initiation, proliferation, progression and metastasis. The location of the neutrophils can be predominantly at the tumor periphery initially, and later within the tumor with different phenotypes initially anti-tumorigenic (N1) and later pro-tumorigenic (N2). Cancer associated fibroblasts (CAFs) modulate the expression of CXCL6 and TGF-β through cardiotrophin-like cytokine factor 1 by polarizing the TANs towards the pro-tumoral phenotype (N2). The N2 phenotype form neutrophil extracellular traps (NETs) which are released by a process of cell death called NETosis[20]. These NETs support tumor growth and increase invasiveness through activation of TLR-4/9-COX2. N2 TANs inhibit the activation or migration of neutrophils into the tumor through the PD-1/PD-L1 pathway[21]. Expression of CD66b, PD-L1, CCL-2, CXCL8, TNF-α, and elevation of CD66b+neutrophils in the peritumoral region has shown decreased survival in HCC patients. Many studies have shown blocking NETs (by inhibiting COX2, inhibiting NETosis by inhibiting cathepsin G) decreased invasion and metastasisin vitro[22]. Studies have shown that TANs cause recruitment of macrophages and Treg cells within the tumor by secreting CCL-2 and CCL-17, resulting in resistance to sorafenib[23]. TME neutrophils act as a principal source for the production of prometastatic Oncostatin M and matrix metalloproteinase which promote angiogenesis by releasing pro-angiogenic factors leading to migration of cancer cells. Evasion of autophagy or delay in apoptosis of neutrophils in the TME is also associated with tumor growth and angiogenesis[24]. The extensive role of TANs reveals new horizons in our understanding of the cancer microenvironment and potential therapeutic options.
DCs-the initiator: DCs are unique cells for capturing pathogens or antigens from tumor cells and presenting them to naive T cells which leads to their differentiation into effector T cells marking the initiation of immune response. Based on the stage of differentiation and development, physiological and pathological environment, DCs are divided into (1) Conventional DCs (cDCs) also known as myeloid DCs (CD141+/CD14- type 1 cDCs and CD1c+/CD14- type 2 cDCs); (2) Plasmacytoid DCs (pDCs) (CD303+ CD304+, secreting type I IFN); and (3) Inflammatory DCs[25]. The interaction of DCs with other immune cells occurs in a sequential manner; DCs presenting antigen to CD4+Th cells through MHC class II and CD8+T cells through MHC class I, which results in a co-stimulatory molecular interaction leading to cytokine production that stimulates CD8+T cells differentiation and expansion[26]. Studies have observed that in patients with HCC, there is lowered expression of co-stimulatory molecules and decreased levels of cDCs and pDCs making the TME appropriate for tumor growth. In HCC, the presence of BDCA2+pDCs increase infiltration of T regulatory cells, which secrete IL-10, and IL-17 producing cells into the tumor. In addition, pDCs and tumor cDCs express Gal9 (ligand of TIM3), PD-L1, MHC-II (for LAG3), and CD80 (for CTLA 4) inducing an immunosuppressive environment in the TME. Newer subsets of DCs (DC-c1-CD1C, DC-c3-CLEC9A, and DC-c4-LAMP3) have been found in treatment naive HCC patients with LAMP3+DCs having a strong association with exhaustion/regulation of T cells.
The TME also diverts the process of dendropoiesis (DCs generation) and tends to polarize the phenotype of DCs which creates an immunosuppressive environment by acting against anti-tumor immunity[27]. Anti-tumor immunity enhancing strategies such as DC based vaccines or immunotherapies are under clinical trials and have shown better outcomes and an enhanced CD4+T cells/CD8+T cells ratio[28]. The profound impact of DCs on immune modulation may lead to the development of new immunotherapies.
Tumor associated macrophages - a double edged sword:Liver parenchyma has a high macrophage density. Cytokines influence macrophage differentiation into classically activated M1 (CD86+) macrophages performing pro-inflammatory functions and M2 (CD163+, CD206+) macrophages which suppress the immune system and perform tissue repair. Liver tumor associated macrophages (TAMs) are commonly associated with CD68+as their marker[29]. In HCC, studies have shown that joint analysis of high level CD206+M2 macrophages and low level CD86+M1 macrophages is associated with aggressive HCC phenotype thus indicating their utility as a prognostic tool for HCC[30]. TAMs promote metastasis, antitumor immunity suppression, angiogenesis and drug resistance. The TME contains two different TAMsi.e.resident macrophages and infiltrating macrophages. Osteopontin/CSF1/CSF1R pathways are other mechanisms leading to the infiltration of macrophages and drug resistance[31]. Activation of M2 macrophages through the Wnt/β-catenin pathway may pose an increased risk for tumor progression in HCC. TAMs modulate the tumor structure, migration, invasion and metastasis through various cytokines such as IL-6, IL-10, TNF-α, VEGF and other signals inhibiting T cells, NK cells cytotoxicity, and differentiation of Tregs. Studies have documented an association between high levels of TAMs in the peri-tumoral region and poor prognosis of HCC[32]. TAMs with actions of M1 macrophages cause immune activation, phagocytosis, and apoptosis of tumor cells. Many newer immune combination therapies targeting these immune suppressive mechanisms are under trials.
Monocytes and Myeloid derived suppressor cells:Recruited through tumoral CCL-2 production, monocytes have antitumoral effects in the early stages of HCC and later the tumor cells evade monocyte induced death and cause progression of the tumor. In the TME monocytes are classified as CD14+monocytes, CCR1+CD14+monocytes, and Myeloid derived suppressor cells-M type. In advanced stages of HCC, CD14+monocytes due to the expression of PDL1/2+, IL-10, and CCL-1 promoted an immunosuppressive environment in the TME[33]. The CCL-15 chemokine recruits the suppressive phenotype of monocytes and promotes immune escape of HCC by increased angiogenesis and metastasis[34].
Monocytes and Myeloid derived suppressor cells (MDSCs) are immature immune cells that suppress the antitumor immunity in tumors. Phenotypically, MDSCs are classified into two types - polymorphonuclear (PMNs) MDSC and monocytic MDSC[35]. They differ in their mechanism in which they mediate the immune suppression. PMN-MDSCs mediate through PGE2, arg-1, and ROS while M-MDSCs facilitate their action through immunosuppressive cytokines (IL-10 and TGF-β), nitric oxide and immunomodulatory molecules such as PD-L1. In HCC, the proportion of M-MDSCs is high, inhibiting NK cell cytotoxicity and inducing Tregs[36]. The TME in HCC is also modulated by MDSC through the production of angiogenic factors and other enzymes promoting angiogenesis and growth of the tumor.
T regulatory cells-suppressors of anti-tumor immunity:Tregs are a specialized subset of T lymphocytes having a distinctive role in the suppression of excessive immune response and mitigating inflammation. Tregs are classified into (1) Natural Tregs possessing (nucleus FOXP3, CD25 and CTLA-4 surface markers); and (2) Induced Tregs (FOXP3 and CD4+ markers)[37]. These cells modify the T cell activation, differentiation, proliferation and function of effector T cells by various genetic mechanisms. Infiltration of Tregs into the TME is influenced by chemokines CCL-17, 22 through the CCR4 receptor. Most CCR4+ Tregs are more immunosuppressive than CCR4- Tregs[38]. Many studies have shown that although Checkpoint inhibitors show a good response in HCC, a few individuals with resistance to immune therapy can be attributed to Treg cells (Figure 2).

Figure 2 lmmune suppression in the tumor microenvironment. An immunosuppressive environment is brought about by the interaction of various immune cells in the tumor microenvironment (TME). N2 pro-tumorigenic tumor associated neutrophils (TANs) influence Tregs and T cells by various chemokines such as CCL-2, CCL-17 and interleukin (IL)-8, tumor necrosis factor, respectively. M2 tumor associated macrophages modulate the TME by influencing Tregs, natural killer (NK) cells and T lymphocytes through IL-6, vascular endothelial growth factor and IL-10, respectively. M type myeloid derived suppressor cells inhibit the NK cells and influence Tregs through IL-6. Cancer associated fibroblasts modulate the effect of N2 TANs via IL-6. Dendritic cells also play a role in the TME by regulating Tregs. CAFs: Cancer associated fibroblasts; PD-1: Programmed death receptor-1; PD-L1: programmed death receptor ligand-1; pDC: Dendritic cell; M-MDSC: Monocytic myeloid derived suppressor cells; NK cell: Natural killer cell; VEGF: Vascular endothelial growth factor; TAN: Tumor associated neutrophils; TAM: Tumor associated macrophages; Tregs: Regulatory T cells; IL: Interleukin.
CAF:These are Fibroblast-specialized cells with a role in the synthesis and maintenance of the extracellular matrix. CAFs are derived from mesenchymal lineage and contribute to tumor promoting inflammation and fibrosis. CAFs can differentiate from blood vessels, epithelial cells, pericytes, adipocytesviaendothelial to mesenchymal transition[39]. In HCC, these fibroblasts can differentiate from cancer cells or vascular cells or from mesenchymal stem cells in bone marrow. Based on the expression of α-smooth muscle actin (α-SMA) and IL-6, two major phenotypes of CAFs are present (1) Myofibroblastic (myCAF); and (2) Inflammatory type (iCAF). The myCAF are more matrix-secreting, TGF-β-responsive with high α-SMA expression, and low-cytokine IL-6 and IL-11 production, and are localized in dense stroma near cancer cells. The iCAF exhibit high IL-6 and IL-11 production with low α-SMA expression and are localized in stroma away from cancer cells[40,41].
CAFs induce changes in the tumor by (1) Angiogenesis through production of VEGF, platelet derived growth factor and CXCL-12; (2) Invasion and metastasis; (3) Immune modulation by recruitment of immune suppressors and suppressors of anti-tumor immunity; and (4) Resistance to therapeutic modalities. CAFs in the TME shape the milieu of the tumor by generating pro-inflammatory cytokines including IL-1β and IL-6 and by expressing ligands CXCL12 and CXCL1 resulting in tumor promotion. The interaction of CAFs with other immune cells such as T cells, NK cells, MDSCs, DCs, TANs, and TAMs in the TME result in immunosuppression. The CAFs have a key role in promoting carcinogenesis of epithelial cells and inducing the generation of MDSC through the IL-3/STAT3 axis and SDF-1α which suppress antitumor immunity[42,43].
Cold and hot tumors concept
Based upon the inflammatory milieu of the TME, no T cell infiltrate, presence of regulatory cytokines, no PD-L1, and increased CAFs, increased MDSC population, the tumor is labeled as “COLD” and poorly responsive to immunotherapy[44]. On the other hand, increased T cell infiltrate, pro-inflammatory cytokines, high PD-L1, increased CD8+T cells, and increased TAMs, the tumor is labeled as “HOT” and is amenable to immunotherapy (Figure 3).
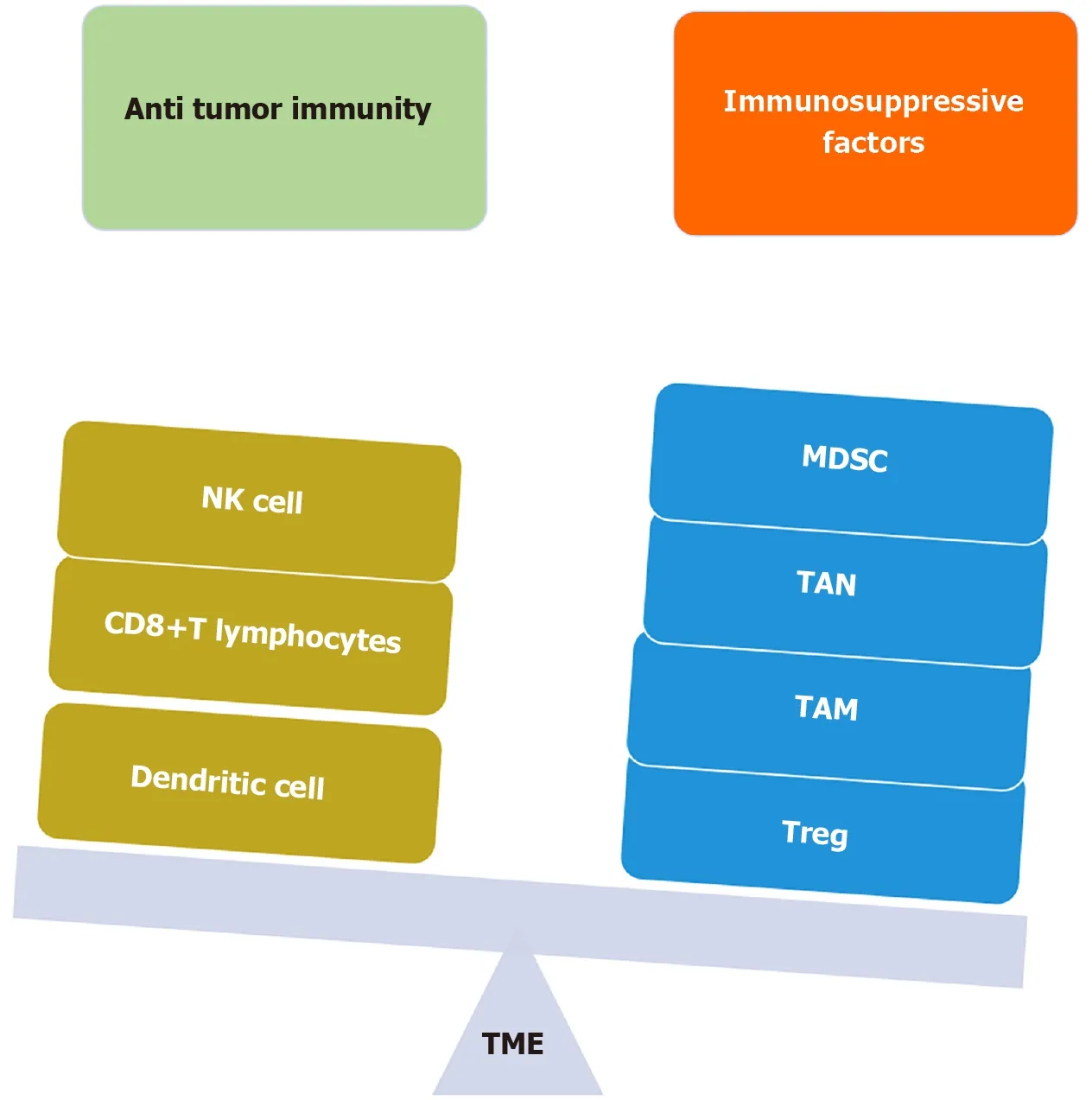
Figure 3 Tumor microenvironment balance. Depicted is the inclination of the tumor microenvironment towards an immunosuppressive environment and its components. NK cell: Natural killer cell; TAN: Tumor associated neutrophils; TAM: Tumor associated macrophages; Treg: Regulatory T cells; MDSC: Myeloid derived suppressor cells; TME: Tumor microenvironment.
lMMUNOTHERAPY lN HCC
The primary treatment options for each HCC stage depends not only on the stage but also on the patient characteristics and profile of the patient. According to BCLC update 2022, systemic therapy is treatment of choice in patients with advanced stage (BCLC-C) HCC and in patients with stage A and B where other treatment options are not feasible or failed[3]. Immune therapy utilizes the body's natural defense mechanisms to combat tumor cells in any cancer. Immune checkpoints (ICs) are molecules present on lymphocytes which regulate the functions of T lymphocytes and influence tumor autoimmunity. Immune cells such as T cells, NK cells, and Tregs express PD-1 checkpoint molecules whereas stromal cells, myeloid cells, and tumor cells express PD-L1/PD-L2 which inhibit the functions of effector T cells and create an immunosuppressive environment. ICIs target these molecules expressed on immune cells to enhance autoimmunity in the TME.
Current status
Atezolizumab (Atez) is a monoclonal antibody against PD-1 and in combination with Bevacizumab (Bev) monoclonal antibody against the VEGF receptor has been approved as first-line therapy for advanced stage HCC (BCLC stage C). The IMbrave150 study[45] showed that the combination arm (Atez 1200 mg/Bev 15 mg/kg) resulted in overall survival (OS) at 12 months of 67.2%vs54.6% in the sorafenib (400 mg BD) arm. Further analysis showed that the Atez/Bev combination resulted in an OS of 19.2 monthsvs13.4 months and progression free survival (PFS) of 6.9 monthsvs4.8 months in the sorafenib arm in unresectable HCC (uHCC), respectively[46]. The beneficial effects of the Atez/Bev combination arm were persistent across BCLC stage B or C, extrahepatic metastases and portal vein invasion. Grade 3 or 4 adverse events related to treatment occurred in 43% and 46% patients in the Atez/Bev combination and sorafenib, respectively. This was a landmark trial and led to FDA approval of this combination (Atez/Bev) in advanced HCC[47]. In a multi-centric retrospective real world evaluation of data, the Atez and Bev combination was well tolerated with no evidence of treatment related deaths or new adverse events across CP-A and CP-B patients with an OS of 14.9 months and PFS 6.8 months[48]. A systematic review of studies from 2002-2020 on systemic therapies in HCC (including all disease stages) examined the association between etiology of HCC and therapy outcomes. The results revealed that immunotherapies were more effective in viral etiologies as compared to non-viral etiologies as compared to TKIs/anti-VEGFs[49]. The viral etiology related HCC is more immunogenic and therefore, ICIs are more effective due to their favorable TME. On the other hand, non-alcoholic steatohepatitis (NASH) related HCC has been shown to accumulate exhausted CD8+PD1+T cells in the TME, and in preclinical models, anti-PD-1 therapy instead of tumor regression led to tumor progression in terms of size as well number of nodules[50]. Another systematic review[51] showed the non-inferiority of LEN to Atez/Bev in achieving an objective response rate (ORR) and disease control rate (DCR) in advanced HCC; however, data were insufficient for evaluation of OS.
The Himalaya trial evaluated the STRIDE regimeni.e.anti-CTLA4 inhibitor tremelimumab single dose (T300 mg) and anti-PD-L1 durvalumab (D1500 mg every 4 wk) in uHCC and found a significant increase in median OS by 2.5 months (16.4 monthsvs13.8 months) as compared to sorafenib (400 mg BD) alone. They showed that a single priming dose of T was sufficient to enhance the efficacy of D in uHCC patients with no increased adverse drug events[52]. This groundbreaking trial led to the recommendation of tremelimumab and durvalumab as first-line therapy for uHCC.
If first-line therapies are not feasible or contraindicated for some reason, monotherapy with TKIs sorafenib/LEN or durvalumab (anti PD-L1) can be considered.
The KEYNOTE 224 phase II trial[53] documented the antitumor activity and safety of pembrolizumab therapy in patients with HCC previously treated with sorafenib. Subsequently, the multi-centric KEYNOTE 240 phase II trial[54] in HCC patients (previously treated with sorafenib) showed no significant difference in OS and PFS after pembrolizumab therapy. The recent KEYNOTE 394 multi-centric trial[55] from Asia, in HCC patients (post sorafenib or progression/intolerance on sorafenib) demonstrated significantly increased OS (14.6 monthsvs13 months,P= 0.01) and PFS (2.6 monthsvs2.3 months,P= 0.003) in the pembrolizumab group compared with placebo, respectively.
The CheckMate 040 phase I/II, non-comparative trial[56] showed safety data in patients with advanced HCC treated with nivolumab (anti-PD-1 inhibitor). The phase 3 trial CheckMate 459 compared nivolumab and sorafenib in advanced HCC and both groups had similar OS and PFS with no significant differences. The CheckMate 040 phase III RCT[57] showed improved ORR and durable response with the combination of nivolumab and ipilimumab therapy in advanced HCC (Table 1).
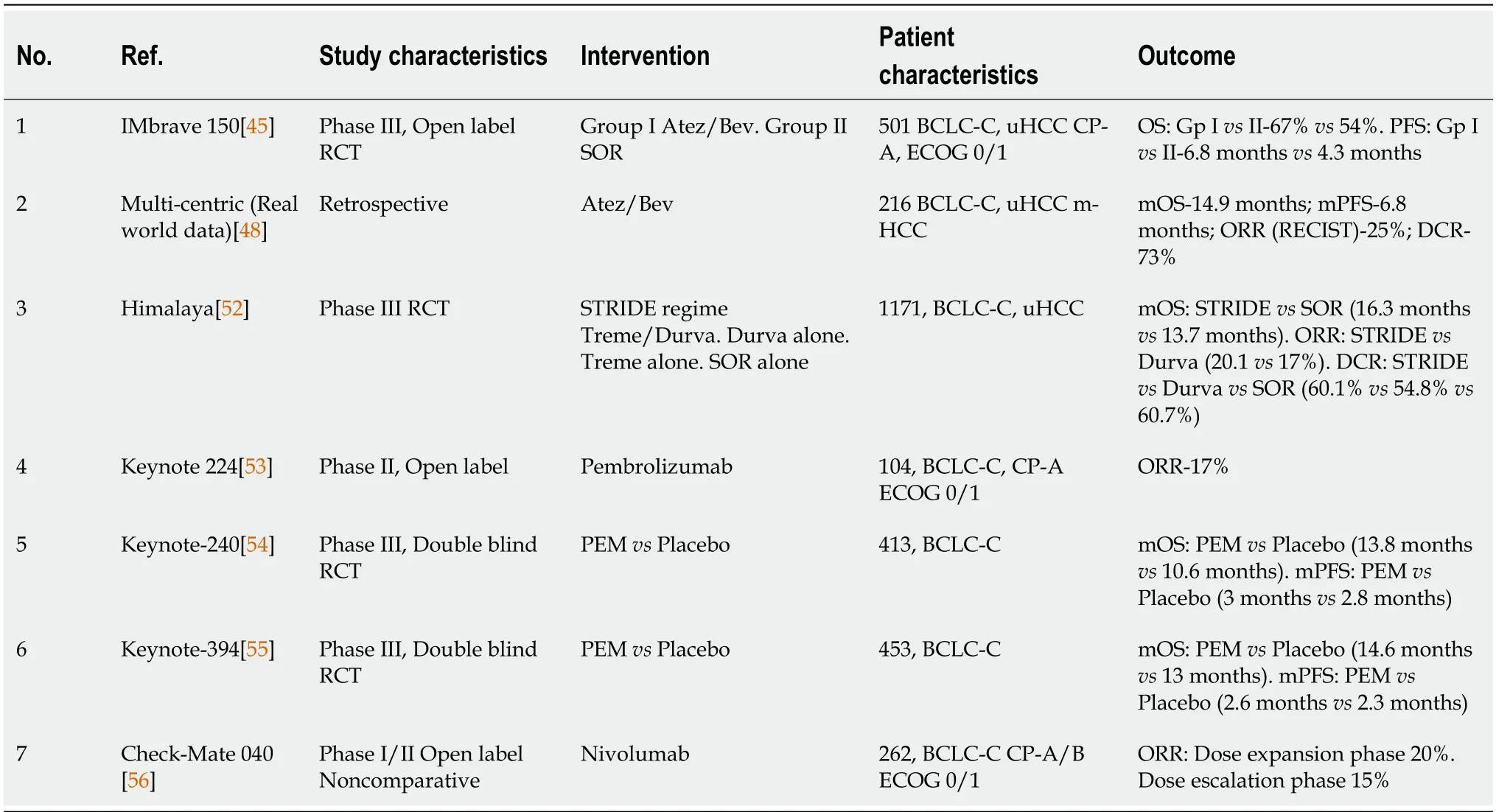
Table 1 Clinical studies on immunotherapy
Predictors of response to immunotherapy
Pre-existing immunityi.e.intra-tumoral CD8+Tcell density, high expression of CD274, low Treg to T effector cell ratio, low expression of oncofetal genes such as GPC3 and AFP, high expression of VEGF receptor 2 and myeloid inflammation signatures are predictors of response to the Atez/Bev combination regimen. Viral etiology related HCC is more immunogenic and therefore, is more responsive to ICIs.
Combination therapies
HCC is a complex tumor where multiple factors such as size of the primary tumor, intrahepatic spread, vascular invasion and metastatic disease need to be addressed. Additionally, liver dysfunction and its severity affect the feasibility of locoregional as well as systemic therapies. The combination of locoregional and systemic therapies has been evaluated in recent trials to improve overall outcomes (Table 2).

Table 2 Clinical studies on combination therapies of immunotherapy with locoregional/tyrosine kinase inhibitors
ICI and transarterial chemoembolization
A propensity score matched study compared the combination of transarterial chemoembolization (TACE) and Atez/Bev against the combination of TACE and LEN (LEN-TACE). They found that both groups showed comparable safety and efficacy in uHCC patients[58]. Another recent Chinese study[59] investigated the combination of TACE and Atez/Bev which resulted in significantly better ORR, OS and PFS as compared to Atez/Bev. The rationale for this, is that TACE decreases the primary tumor load and therefore, the burden of immunosuppressive Treg cellsetc.and induces hypoxia in the TME. Therefore, CTLs increase in the TME and hypoxia induces an increase in VEGF expression and ICIs (Atez/Bev) have better action due to favorable conditions in the TME. For BCLC stage B, the recommended treatment modality is TACE. Switching to ICIs before deterioration of liver function in patients with BCLC stage B could improve their prognosis and survival. The REPLEC study[60] included HCC patients with BCLC stage B beyond up to seven criteria (unsuitable for TACE) UT-7/multiple/Child Pugh A treated with Atez/Bev who showed an ORR and DCR of RECIST and mRECIST of 17.7%/84.7% and 42.5%/86.2%, respectively.
ICIs and TKIs
The phase 1 KEYNOTE-524 trial[61] demonstrated that the LEN + pembrolizumab (PEM) combination resulted in a median PFS of 9.3 months, ORR of 46% by mRECIST, and median OS of 22 months in patients with uHCC in 29% of BCLC stage B (not suitable for TACE) and in 71% stage C patients. The rationale behind this combination was that LEN, due to its immunomodulatory effects, inhibits proangiogenic and immunosuppressive mechanisms in the TME and enhances the antitumor effects of pembrolizumab. However, the recent LEAP-002 phase 3 trial[62] failed to show any benefit of LEN + PEM combination therapy in uHCC.
ICIs and Ablation
A recent study demonstrated that tremelimumab (anti-CTLA4 Ab) combined with ablation achieved good anti-tumor activity due to enhanced CD8+T cells in the tumor periphery after ablation[63].
ICIs and surgical resection of HCC
Surgical resection in HCC is confined to BCLC very early stage and early stage of HCC (stage A). In advanced stages of uHCC, ICIs are used as bridging therapy for tumor downstaging, negative selection and as neoadjuvant therapy[64]. Downstaging of HCC refers to a shift in tumor stage to a lower level after immunotherapy when surgical intervention can be considered. Negative selection refers to a new concept of “absence of appearance of new lesions after immunotherapy with steady response.” This can be treated as localized disease and surgical options can be tried as definitive management. Neoadjuvant therapy is used to shrink the tumor size and allow wider safety margins during surgery. Neoadjuvant immunotherapy is administered for patients with either early resectable tumor or initially unresectable tumor for downstaging[65].
In a study involving 54 patients with uHCC, who received combination immunotherapy followed by radical surgery, pathological evaluation of postoperative specimens confirmed 21.4% (n= 3) of patients achieved a complete response and 78.6% (n= 11) achieved PR[66]. Zhanget al[67] reported 10 patients with HCC and major vascular invasion who achieved a 12-month recurrence-free survival rate of 75% after combinations of ICI and TKI with subsequent salvage surgery[67].
Immune therapy related adverse events
Tolerance of the immune system is the ability to prevent an immune response against a particular antigen. Immune therapy (ICIs) breaks the tolerance of the body’s immune system which produces immune related adverse events. Based on common terminology criteria for adverse events grading, the severity of immune therapy related adverse events (irAEs) are Grade 1 mild, Grade 2 moderate, Grade 3 severe, Grade 4 life threatening, Grade 5 death[68]. Inhibition of checkpoint molecules which prevent the tumor cells escaping the immune system can cause disruption in tolerance of the immune system (mainly peripheral T cells) leading to proliferation of immune cells and high inflammation and autoimmunity. Hence, most common sites involved would be skin and colon as they predominantly depend on peripheral T cell tolerance for maintaining immune homeostasis[69]. Other mechanisms of irAEs involve autoreactive B cells, cytokines and other host factors.
In HCC patients receiving ICIs, the incidence of irAEs is not higher than that in other carcinomas. The most common irAEs are skin manifestations, followed by gastrointestinal effects such as diarrhea. Hepatic irAEs include raised liver enzymes; however, they are grade 1 to 3 and are not life threatening. Patients with hepatitis B or C seropositive status are not prone to developing a flare if antiviral therapy is started and continued during ICI administration. However, CP-B patients due to their underlying severe liver dysfunction are more prone to severe irAEs. Nivolumab and pembrolizumab monotherapy in HCC patients resulted in rash and pruritus, which were the most common manifestations with an incidence of 11%-23% and 13%-19%, respectively. The incidence of colitis was 1% in patients treated with PD-L1 antibodies and 2.6% in patients treated with the combination of PD-L1 antibody and CTLA-4 antibody. Pneumonia was the main irAE with an incidence of 3% in patients treated with nivolumab[70]. Following tremelimumab therapy, grade 3 or higher encephalopathy was observed, but this may have been attributed to underlying cirrhosis than immunotherapy. Hypertension was observed to be the most common adverse event in patients treated with Atez/Bev with an incidence of 29.8%. Hypertensive encephalopathy, nephrotic syndrome, bleeding, myelosuppression and infection are severe irAEs in patients receiving Atez/Bev and these are influenced mostly by Bev. There is a need for vigilant monitoring to identify adverse events related to immunotherapy and prompt intervention is required for optimal patient outcomes.
CHALLENGES WlTH lCls
Tumor resistance to ICIs is a major challenge which is multifactorial and includes the following: (1) HCC mutational burden-total somatic mutations in HCC responsible for immune cells regulation; (2) Genetic pathways[71] (overactivation of beta-catenin) leading to decreased CD8+T cells infiltration and low PD-L1 expression in the TME; (3) TP53 inactivating mutations leading to ICIs resistance and tumor progression[72]; and (4) T cell exhaustion due to interaction of LAG 3 molecules and overexpressed FGL-1 in the TME[73].
CONCLUSlON
ICIs have resulted in a paradigm shift in the management of advanced HCC. However, there is still a long way to go. There is a need to evaluate ICIs use in early HCC and to evaluate their role in downstaging of tumors for curative therapies such as resection or liver transplantation. Future strategies regarding other targets may overcome the ICI resistance seen in clinical practice. With upcoming NASH and obesity epidemics and NASH-HCC being less immunogenic with ICI resistance, it is necessary to determine how this low immunogenicity may be converted or reverted back to the immunogenic state to achieve ICI response. Cell based therapies or vaccines are other areas requiring research.
FOOTNOTES
Author contributions:Gupta T wrote the paper and critically analyzed the manuscript; Jarpula NS collected the data and literature and drafted the manuscript.
Conflict-of-interest statement:No conflict of interest.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:India
ORClD number:Tarana Gupta 0000-0003-3453-2040.
Corresponding Author's Membership in Professional Societies:Indian National Association for Study of Liver Diseases, No. 1319; American Association for The Study of Liver Diseases, No. 226223.
S-Editor:Fan JR
L-Editor:Webster JR
P-Editor:Guo X
 World Journal of Hepatology2024年3期
World Journal of Hepatology2024年3期
- World Journal of Hepatology的其它文章
- Update in lean metabolic dysfunction-associated steatotic liver disease
- Retrospective study of the incidence, risk factors, treatment outcomes of bacterial infections at uncommon sites in cirrhotic patients
- Palliative long-term abdominal drains vs large volume paracenteses for the management of refractory ascites in end-stage liver disease
- Comprehensive prognostic and immune analysis of sterol Oacyltransferase 1 in patients with hepatocellular carcinoma
- Prediction model for hepatitis B e antigen seroconversion in chronic hepatitis B with peginterferon-alfa treated based on a responseguided therapy strategy
- lnfluence of nonalcoholic fatty liver disease on response to antiviral treatment in patients with chronic hepatitis B: A meta-analysis