
SIRT3缺陷对脓毒症心肌损伤的影响及作用机制
2024-05-06 09:38:30徐天华刘鹏昊崔德荣
医学研究杂志 2024年3期
徐天华 刘鹏昊 崔德荣
脓毒症是由宿主对感染的反应失调引起的危及生命的器官功能障碍,是全球危重症患者死亡的主要原因之一,院内病死率可达32.8%,当存在休克时甚至接近40%~60%,每年可能有数十万人死亡[1,2]。脓毒症诱发的心脏功能障碍与患者的不良结局和更高的病死率相关,是脓毒症的主要并发症之一[3,4]。目前关于脓毒症心肌损伤的具体发病机制仍然存在争议,其中微循环障碍被认为是脓毒症心肌损伤的重要环节,微血管内皮损伤可引起心肌灌注减少和组织供氧不足,导致心力衰竭[5,6]。在缺氧、炎症等条件刺激下,内皮细胞获得间充质细胞的某些特征并逐渐失去内皮性质,这一过程被称为内皮-间充质转化(endothelial-to-mesenchymal transition, EndMT),其导致细胞外基质成分(如胶原蛋白和纤连蛋白)积累,引起内膜增生和血管狭窄,进而参与了许多疾病的纤维化过程[7,8]。自噬作为细胞应对外界刺激的一种重要生理机制,能够清除受损的细胞组分进而维持细胞稳态。既往研究表明,自噬可通过影响EndMT保护缺氧条件下的人心脏微血管内皮细胞[9]。然而,有关EndMT在脓毒症心肌损伤中的作用机制报道较少。
SIRT3(sirtuin 3)主要位于线粒体中,通过对多种蛋白质的去乙酰化修饰维持线粒体正常的生物学功能,其活性与自噬、氧化应激等多种生理通路有关[10,11]。研究表明,SIRT3可通过激活Foxo3a、AMPK和SOD2调节自噬过程,进而减轻心肌缺血再灌注损伤[12]。然而SIRT3在脓毒症心肌损伤中的具体调控机制尚不明确。因此本研究拟采用野生型(wild type, WT)小鼠与SIRT3基因敲除(SIRT3-/-)小鼠制备脓毒症心肌损伤模型,观察SIRT3缺陷对脓毒症心肌损伤的影响,探究其作用机制,为临床治疗脓毒症心肌病提供新的思路。
材料与方法
1.材料与试剂:兔抗小鼠SIRT3抗体,兔抗小鼠血小板-内皮细胞黏附分子(platelet-endothelial cell adhesion molecule, CD31)抗体,兔抗小鼠微管相关蛋白1A/1B-轻链3(microtubule-associated protein 1A/1B light chain 3,LC3)单克隆抗体,小鼠抗小鼠泛素结合蛋白p62(sequestosome 1,SQSTM1/p62)抗体,小鼠抗小鼠α-平滑肌肌动蛋白(α-smooth muscle actin, α-SMA)抗体,Alexa Fluor®546标记的山羊抗兔IgG H&L抗体和Alexa Fluor®488标记的山羊抗小鼠IgG H&L抗体均购自英国Abcam公司,脂多糖(lipopolysaccharide, LPS)购自美国 Sigma 公司。人心脏微血管内皮细胞及内皮细胞专用培养基购自上海中乔新舟公司,SIRT3过表达慢病毒及阴性对照病毒购自上海吉凯基因公司,增强型化学发光显影剂(enhanced chemiluminescence, ECL)购自南京诺唯赞生物科技有限公司,RIPA组织蛋白裂解液购自美国Thermo Fisher公司,蛋白定量试剂盒(BCA protein assay kit)购自上海碧云天生物技术股份有限公司。
2.动物模型制备及分组:本研究符合《美国NIH实验室动物使用指南》的规定, C57BL/6野生型小鼠购自上海雷根生物科技有限公司[实验动物许可证号: SCXK(苏)2020-0009],SIRT3基因敲除小鼠(上海交通大学医学院附属瑞金医院实验室赠予)饲养于上海市第六人民医院实验动物中心[实验动物许可证号: SYXK(沪)2021-0028]。参照既往研究方法,随机选取8~12周雄性小鼠腹腔注射LPS(10mg/kg)或相同体积的0.9%氯化钠溶液(normal saline, NS),24h之后收集心脏组织用于检测[13,14]。动物分组设立为WT-NS组、WT-LPS组、SIRT3-/--LPS组、SIRT3-/--NS组,每组8只小鼠。
3.细胞处理及分组:人心脏微血管内皮细胞培养于含5%胎牛血清及1%内皮生长因子的内皮细胞培养基中,放置在正常细胞培养箱中培养。转染时细胞以90000/孔的密度接种在6孔板中,培养16~24h后加入SIRT3过表达慢病毒及阴性对照病毒,感染12h之后换新鲜培养基,继续培养至72h后于荧光显微镜下观察病毒转染效率。取生长状态良好的人心脏微血管内皮细胞,经LPS(10μg/ml)刺激24h之后收集细胞蛋白进行检测。细胞分组设立为对照组、LPS组、LPS+SIRT3过表达组、阴性对照病毒组。
4.心脏超声:小鼠LPS腹腔注射24h后,经异氟烷吸入麻醉诱导并固定于恒温板上。超声探头放置于胸骨旁左心室切面, 探头频率为30MHz,探测深度为13mm,待小鼠心率与呼吸稳定后,记录并分析M模式下小鼠收缩末期及舒张末期左心室后壁厚度、左心室舒张末期容积。
5.天狼星红染色:4%多聚甲醛固定心脏组织,常规石蜡包埋及切片。石蜡切片脱蜡至水,放入天狼星红染液中染色8min,用无水乙醇脱水后放入干净的二甲苯中透明5min,中性树胶封片。染色后在光学显微镜下进行观察,正常心肌组织呈黄色,而胶原组织呈红色。使用胶原组织面积占该视野中总面积的百分比进行统计,分析心脏纤维化水平和胶原蛋白的空间分布情况。
6.Western blot法检测:动物模型制备成功后,取部分心肌组织于冰上裂解,采用BCA法测定蛋白浓度。样品经常规电泳、转膜之后,使用脱脂牛奶室温封闭1h。将PVDF膜放入按照1∶1000稀释的 CD31、α-SMA、LC3、p62、SIRT3和1∶5000稀释的α-tubulin和甘油醛-3-磷酸脱氢酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)抗体中,置于摇床上4℃孵育过夜,清洗之后加入稀释的二抗,室温下孵育1h。ECL显影,凝胶成像系统进行拍照。GAPDH和α-tubulin作为内参蛋白,目标蛋白与内参的比值作为蛋白的相对表达量。
7.免疫荧光:石蜡切片脱蜡至水,置于盛满EDTA抗原修复缓冲液(pH值为8.0)的修复盒中于微波炉内进行抗原修复。用含10%胎牛血清及0.3% Triton X-100的封闭液室温封闭1h,随后加入一抗稀释液,兔抗小鼠CD31抗体(1∶200),小鼠抗小鼠α-SMA抗体(1∶400),放于湿盒中4℃孵育过夜。加入Alexa Fluor®546标记的山羊抗兔IgG H&L抗体(1∶500)和Alexa Fluor®488标记的山羊抗小鼠IgG H&L抗体(1∶500),室温避光孵育1h, 加入含核酸染料4′, 6-二脒基吲哚(4′, 6-diamidino-2-phenylindole, DAPI)的封片剂进行封片,共聚焦显微镜下采集图像并分析。

结 果
1.SIRT3缺陷对LPS刺激下小鼠心脏结构和功能的影响:如图1所示,心脏超声观察结果表明,相对于WT-NS组和WT-LPS组,SIRT3-/--LPS组的左室后壁厚度明显增加,且左心室舒张末期容积显著降低(P<0.001)。而WT-LPS组与WT-NS组比较,差异无统计学意义。
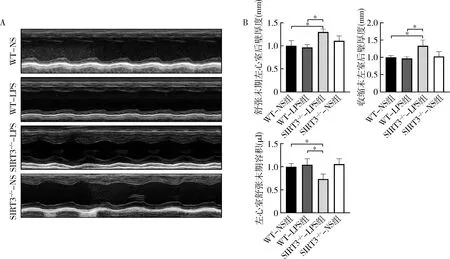
图1 SIRT3缺陷对LPS刺激下小鼠心脏结构和功能的影响(n=5)
2.SIRT3缺陷对LPS刺激下小鼠心脏纤维化的影响:天狼星红染色结果显示(图2),与WT-NS组比较,WT-LPS组的胶原蛋白含量明显增多,且主要沉积于血管周围(P<0.01)。而SIRT3-/--LPS组胶原蛋白沉积程度进一步加深(P<0.001)。

图2 SIRT3缺陷对LPS刺激下小鼠心脏纤维化的影响(n=4)
3.SIRT3缺陷对LPS刺激下小鼠心脏微血管内皮细胞的影响:使用免疫荧光技术检测心脏CD31、α-SMA蛋白表达情况(图3),结果显示,与WT-NS组比较,WT-LPS组血管内皮细胞标志蛋白CD31荧光强度降低(P<0.01),间充质细胞标志蛋白α-SMA荧光强度升高(P<0.05),表明EndMT的发生;而SIRT3-/--LPS组较WT-LPS组CD31荧光强度更低(P<0.05),α-SMA荧光强度更高(P<0.001),说明SIRT3-/--LPS组的内皮-间充质转化程度更重。
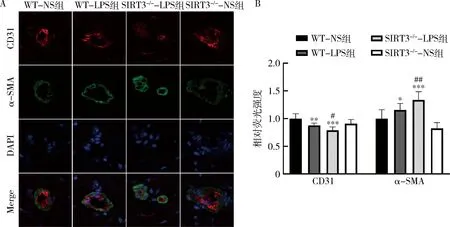
图3 SIRT3缺陷对LPS刺激下小鼠心脏微血管内皮细胞的影响(n=4)
4.SIRT3缺陷对LPS刺激下小鼠心脏组织自噬水平的影响:使用Western blot法检测心脏LC3、p62、SIRT3的蛋白表达情况(图4),结果显示,与WT-NS组比较,WT-LPS组自噬水平明显增加,LC3-Ⅱ蛋白表达水平升高(P<0.001),p62蛋白表达水平降低(P<0.01),且WT-LPS组SIRT3蛋白表达明显下降(P<0.05)。而SIRT3-/--LPS组较WT-LPS组自噬水平明显下降:LC3-Ⅱ蛋白表达显著降低(P<0.05),p62蛋白表达升高(P<0.001)。
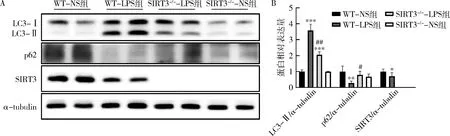
图4 SIRT3缺陷对LPS刺激下小鼠心脏组织自噬水平的影响(n=4)
5.SIRT3过表达对LPS诱导的人心脏微血管内皮细胞间充质转化及自噬水平的影响:使用Western blot法检测人心脏微血管内皮细胞LC3、p62、SIRT3、CD31、α-SMA的蛋白表达情况(图5),结果显示,与对照组比较,LPS组自噬应激性增强,LC3-Ⅱ蛋白表达水平升高(P<0.001),SIRT3蛋白表达明显下降(P<0.001),并伴随EndMT的发生,CD31蛋白表达量下降(P<0.01),α-SMA蛋白表达量增加(P<0.05)。而SIRT3过表达导致人心脏微血管内皮细胞自噬水平进一步升高:LC3-Ⅱ蛋白表达显著升高(P<0.05),p62蛋白表达降低(P<0.01),且抑制了LPS诱导的EndMT水平,CD31蛋白表达量升高(P<0.001),α-SMA蛋白表达量下降(P<0.001)。
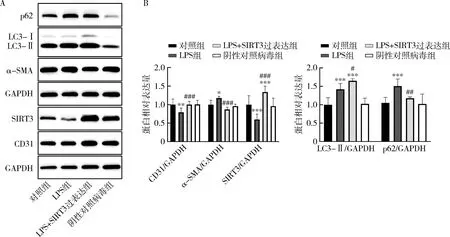
图5 SIRT3过表达对LPS诱导的人心脏微血管内皮细胞间充质转化及自噬水平的影响(n=4)
讨 论
心脏功能障碍是脓毒症相关心血管衰竭的关键特征,与临床预后密切相关,然而脓毒症诱发心肌损伤的治疗手段有限,其病理生理机制仍不明确[15]。研究表明,脓毒症可导致心脏纤维化并影响患者的长期生存[3,16]。以胶原纤维过度沉积为特征的心脏纤维化会改变心肌结构,损害收缩和舒张功能并逐渐进展为心力衰竭,心脏纤维化的严重程度与心脏病患者较高的长期病死率有关[17]。本研究发现,LPS可诱导野生型小鼠心脏SIRT3蛋白表达量下降,且心脏超声和天狼星红染色的结果表明,与WT-NS组比较,LPS刺激可导致心脏胶原蛋白含量增加,且SIRT3-/--LPS组增加更显著。相较于WT-NS组,SIRT3-/--LPS组左心室舒张末期容积降低、左心室室壁厚度增加,但WT-LPS组的相关心超指标差异不显著,这可能与SIRT3在LPS诱导的心肌损伤中的保护作用有关,而SIRT3的缺陷加重了LPS诱导的小鼠心脏结构及功能损伤,说明SIRT3的表达量与LPS诱导的心脏功能障碍及纤维化水平有关。
内皮-间充质转化是内皮细胞的表型向间充质细胞变化的过程,包括紧密连接的丧失,运动性增强和细胞外基质蛋白分泌增加,以内皮基因/蛋白质表达减少和(或)间充质基因/蛋白质的表达增加为特征[18]。心脏微血管内皮细胞的内皮-间充质转化可引起心脏纤维化并通过分泌TGF-β1诱导心肌细胞凋亡[19]。本研究结果显示,LPS可诱导野生型小鼠心脏组织中血管旁胶原蛋白沉积增多,血管内皮细胞标志蛋白CD31表达水平下降而间充质细胞标志蛋白α-SMA表达水平升高,且SIRT3-/--LPS组这一变化更加显著。人心脏微血管内皮细胞经LPS刺激后亦会发生EndMT并伴随SIRT3蛋白表达下降,而SIRT3过表达减轻了LPS诱导的EndMT水平。提示LPS诱导的脓毒症小鼠中SIRT3缺陷可促进心脏微血管内皮细胞发生EndMT,并且EndMT的程度与心脏胶原蛋白的含量有关,而增加SIRT3的表达可以抑制LPS诱导的心脏微血管内皮-间充质转化。
自噬可通过清除受损的细胞器及错误折叠或聚集的蛋白质维持细胞稳态。既往研究表明,自噬通量的恢复可抑制活性氧积累导致的内皮-间充质转化,从而改善阿霉素诱导的心脏纤维化[20]。LC3有Ⅰ型和Ⅱ型两种形式,其中LC3-Ⅱ的含量与自噬体形成的程度呈正比[21,22]。p62蛋白与自噬降解水平相关[23]。本研究发现,WT-LPS组小鼠心脏组织LC3-Ⅱ的表达水平升高且p62的表达水平下降,提示LPS可诱导小鼠心脏组织自噬水平应激性升高。而与WT-LPS组比较,SIRT3-/--LPS组小鼠心脏组织自噬水平下降,说明LPS诱导的脓毒症小鼠中SIRT3缺陷可抑制自噬激活。同时人心脏微血管内皮细胞经LPS刺激后自噬应激性增强,而 SIRT3过表达导致自噬水平进一步增加。提示LPS诱导的脓毒症小鼠中EndMT的程度可能与自噬水平相关,且 SIRT3参与自噬水平的调控。
SIRT3在治疗心血管疾病领域具有很大潜力,从中药中提取的一些成分,如白藜芦醇、和厚朴酚等,可通过激活SIRT3发挥对心血管疾病的保护作用[24]。既往关于SIRT3的研究聚焦于其介导的脓毒症心肌细胞损伤,机制主要涉及三磷酸腺苷(adenosine triphosphate,ATP)合成障碍等,而关于SIRT3调控脓毒症心脏微血管内皮细胞损伤的机制研究较少[25~27]。本研究从脓毒症心脏微循环障碍这一角度出发,发现SIRT3缺陷可抑制LPS诱导的自噬激活,参与调控EndMT,进而影响脓毒症心脏功能及其纤维化。因此,笔者推测SIRT3可能作为脓毒症心肌损伤的有效治疗靶点。
本研究存在的不足:①缺乏特异性敲除心脏内皮细胞SIRT3基因的小鼠,有关SIRT3调控内皮-间充质转化的直接证据尚不充分;②动物模型未设置多个时间点进行检测,且未干扰自噬相关蛋白进行分子之间上下游关系的验证,关于SIRT3的调控靶点有待进一步明确,SIRT3与自噬及心肌纤维化之间的因果关系需要更深入的研究加以阐述。
综上所述,本研究发现,LPS诱导的脓毒症小鼠中SIRT3缺陷可降低心脏组织自噬水平,促使心脏微血管内皮细胞发生内皮-间充质转化,进而加重心脏舒张功能障碍及纤维化程度。SIRT3与脓毒症心脏纤维化存在联系,其中的机制有待于进一步探究。
利益冲突声明:所有作者均声明不存在利益冲突。
猜你喜欢
昆明医科大学学报(2021年3期)2021-07-22 07:40:00
昆明医科大学学报(2021年3期)2021-07-22 07:39:10
昆明医科大学学报(2021年5期)2021-07-22 07:32:50
现代临床医学(2021年2期)2021-03-29 05:32:44
世界科学技术-中医药现代化(2021年10期)2021-03-02 05:52:12
中华养生保健(2020年4期)2020-11-16 01:31:40
中国中医急症(2019年10期)2019-05-21 07:20:46
中华老年多器官疾病杂志(2016年9期)2016-04-28 08:52:44
中国医药导报(2015年26期)2015-02-28 22:07:44
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:49
