
Electronic origin of structural degradation in Li-rich transition metal oxides: The case of Li2MnO3 and Li2RuO3
2024-04-23 13:30:18PengZhang
Journal of Semiconductors 2024年4期
Peng Zhang
Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
Abstract: Li2MnO3 and Li2RuO3 represent two prototype Li-rich transition metal (TM) oxides as high-capacity cathodes for Liion batteries, which have similar crystal structures but show quite different cycling performances.Here, based on the first-principles calculations, we systematically studied the electronic structures and defect properties of these two Li-rich cathodes, in order to get more understanding on the structural degradation mechanism in Li-rich TM oxides.Our calculations indicated that the structural and cycling stability of Li2MnO3 and Li2RuO3 depend closely on their electronic structures, especially the energy of their highest occupied electronic states (HOS), as it largely determines the defect properties of these cathodes.For Li2MnO3 with low-energy HOS, we found that, due to the defect charge transfer mechanism, various defects can form spontaneously in its host structure as Li ions are extracted upon delithiation, which seriously deteriorates its structural and cycling stability.While for Li2RuO3, on the other hand, we identified that the high-energy HOS prevents it from the defect formation upon delithiation and thus preserve its cycling reversibility.Our studies thus illustrated an electronic origin of the structural degradation in Li-rich TM oxides and implied that it is possible to improve their cycling performances by carefully adjusting their TM components.
Key words: defects; Li-ion batteries; first-principles calculations
1.Introduction
Over the past three decades, Li-ion batteries have dominated the rechargeable battery technology for powering portable electronic devices, such as cell phones, laptop computers and iPads that have transformed global communications[1].More recently, with the growing demand for secure and sustainable energy supply of modern society, new applications of Li-ion batteries quickly emerge especially for powering electric vehicles and assembling grid energy storage systems for renewable energies[2-4].However, to meet these large-scale applications, it is important to improve the energy density of Li-ion batteries to over 500 Wh/kg, which is still beyond the current Li-ion technology.This is because, essentially, all of today’s Li-ion batteries use intercalation reactions at both electrodes, which are in most cases restricted to the one-electron redox process per transition metal (TM) ion[5].This redox mechanism imposes a fundamental limitation on the capacity of both electrodes, especially the cathodes, of Liion batteries.For instance, the two representative cathodes used today are LiCoO2[6]and LiFePO4[7], both of which show limited capacities of below 200 mAh/g.
Notably, the discovery of Li-rich TM oxides, Li1+xTM1-xO2,as high-capacity cathodes at the beginning of this century may provide a promising way to address this challenge[8-11].It has been demonstrated that materials, such as Li1+xNiyCozMn1-x-y-zO2, can deliver a capacity of over 250 mAh/g, much higher than that of layered TM oxides[8].The excess capacity of Li1+xTM1-xO2is now recognized to come from the anionic redox activity involving O-dominated states, which is beyond the traditional cationic redox chemistry[12-14].However, the real-world application of these highcapacity cathodes is still plagued by critical issues, such as sluggish kinetics, voltage hysteresis, as well as long-term capacity and voltage fade[15-19].These degradations of Li1+xTM1-xO2have been frequently ascribed to their structural changes upon delithiation, associated with the formation of various defects, such as TM migration and lattice oxygen loss[20,21].Nevertheless, the origin of these detrimental structural changes and their precise relationship to the redox chemistry of Li1+xTM1-xO2remains unknown, leaving the question of how to control and improve the performances of these promising cathodes open.Notably, different from their 3dcounterparts, the recent experiments have demonstrated that 4dand 5dLi1+xTM1-xO2, such as Li2RuO3and Li2IrO3, can possess much better cycling reversibility with negligible capacity fade and O2release[22-25].These observations imply that the atomic configuration of TM ions may have significant influences on the structural stability of Li1+xTM1-xO2and thus provide a promising way to improve their cycling performances by adjusting TM components, whereas the underlying mechanism behind this phenomenon is still not fully understood.
To address these challenges, here we performed first-principles calculations to systematically study the electronic structures and defect properties of Li2MnO3and Li2RuO3, which are the representative 3dand 4dLi1+xTM1-xO2, respectively.Our calculations revealed that the structural and cycling stability of these two cathodes depend closely on their electronic structures, especially the energy of their highest occupied electronic states (HOS), from which the compensating electrons would be removed upon delithiation.For Li2MnO3, we found that its HOS are mainly determined by O 2porbitals, thus having a relatively low energy.As a consequence, injection of holes into these HOS upon delithiation would spontaneously trigger the formation of various defects, such as Mn-Li antisites (MnLi), Mn interstitials (Mni), Li interstitials (Lii), and O vacancies (VO), in Li2MnO3, which are detrimental to its structural stability.The underlying mechanism is that, due to the low-lying HOS, the charge transfer from defect levels to the injected holes upon delithiation could gain an energy that is sufficiently large to compensate the energy cost for the defect formation in Li2MnO3.While for Li2RuO3, on the other hand, we found that its HOS have a predominant Ru 4dcomponent that promotes these states to a higher energy than that of Li2MnO3.Consequently, the charge transfer from defect levels to hole states could gain only a small energy that is not sufficient to trigger the defect formation in Li2RuO3.Thus, by carefully comparing these two prototype cathodes, our calculations uncovered an electronic origin for the structural degradation in 3dLi1+xTM1-xO2and meanwhile explained why the cycling reversibility can be significantly improved in their 2pcounterparts.
2.Computational method
The first-principles calculations were carried out by using the projector augment wave (PAW) method[26]within the framework of density functional theory (DFT) as implemented in VASP[27], with the exchange-correlation energy treated by the strongly constrained and appropriately normed (SCAN) functional[28].The wave functions were expanded by using the plane waves up to a kinetic energy cutoff of 520 eV and the Brillouin-zone integration was approximated with a 12 × 12 × 12k-mesh.During structural optimizations, the lattice vectors and atomic coordinates were all fully relaxed until the force on each atom is less than 0.01 eV/Å.The defect calculations were performed by using the DASP code[29]with a supercell of 432 atoms and a gamma-onlykpoint.
3.Results and discussion
The crystal structure of Li2MnO3consists of cubic closedpacked O ions with alternative sheets of octahedral sites between them occupied by Li and [Li1/3Mn2/3], as show in Fig.1(a).If Li and Mn ions in [Li1/3Mn2/3] layers are fully disordered,thenthestructureresemblesthatoflayered LiTMO2with theRm symmetry.However,theexperimentalmeasurements have identified that [Li1/3Mn2/3] layers actually possess a honeycomb-like cation ordering, which leads to the structure of Li2MnO3with a lowered symmetry of C2/m[30].Like Li2MnO3, Li2RuO3also crystalizes into a layered structure,where Li and [Li1/3Ru2/3] layers stack alternatively, giving a formula of Li[Li1/3Ru2/3]O2in the conventional layered LiTMO2notation, as shown in Fig.1(b).However, compared to Li2MnO3, a minor structural difference still exits for Li2RuO3as its unit cell has the C2/c symmetry with a slightly distorted O framework[31].The calculated lattice constants of the unit cells for both Li2MnO3and Li2RuO3are given in Table 1,which show good agreement with the experimental measurements[30,31].
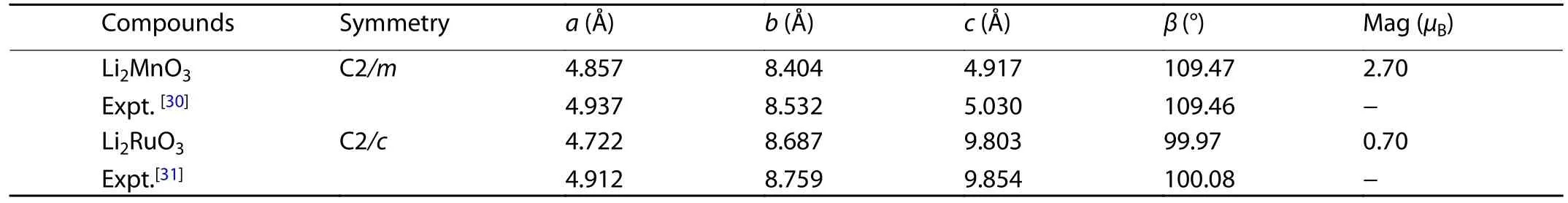
Table 1.The calculated Lattice constants of the unit cells for Li2MnO3 and Li2RuO3, as well as the magnetic moments (Mag) on the Mn and Ru ions.
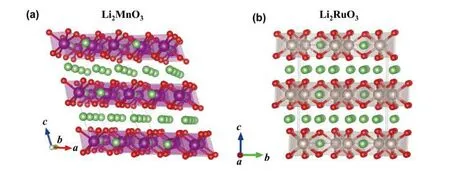
Fig.1.(Color online) The crystal structures for (a) Li2MnO3 and (b) Li2RuO3.The Li, Mn, Ru and O ions are represented by green, purple, light brown, and red balls, respectively.
Fig.2(a) plots the spin-polarized band structure and projected density of states (PDOS) for Li2MnO3.It is clear to see that our calculations with the SCAN functional indicated that Li2MnO3is a semiconductor with a band gap of 2.1 eV, which is in consistent with the previous DFT + U calculations[32].Moreover, from the PDOS, we found a profound hybridization between the Mn 3dand O 2porbitals within a wide energy range from -6 to 5 eV, indicating strong covalent bonds formed between the Mn and O ions.The conduction band minimum (CBM) and valence band maximum (VBM) of Li2MnO3were identified to have the predominant Mn 3dand O 2pcharacters, respectively, which can be further understood by the molecular-orbital diagram, as shown in Fig.2(b).Here, since each Mn ion is octahedrally coordinated with eight O ions, its 3dorbitals would split into the triply degeneratet2gand doubly degenerateegstates, which can further couple with O 2porbitalstoformthebondingandantibondingmolecular orbitals(MOs).ForLi2MnO3,the Mn4+ions havethe 3d3atomic configuration and the high-spin magnetic ground state, as shown in Table 1.Thus, its CBM and VBM correspond to the Mn-dominated and O-dominated MOs, respectively.Based on this band structure analysis, it is natural to expect that, as Li ions are extracted from Li2MnO3upon delithiation, the compensating electrons would be removed from the O-dominated MOs, thereby suggesting an anionic redox process, in accordance with the experimental observations[9].However, it should be noted that injection of holes into O 2porbitals may not be stable, since it usually leads to the formation of compensating defects[33,34].
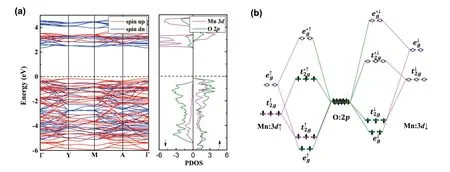
Fig.2.(Color online) (a) The band structure and projected density of states for Li2MnO3 and (b) the corresponding molecular-orbital diagram.
To check the structural stability of Li2MnO3upon delithiation, its defect properties were calculated, following the standard defect calculation method[29,34].Fig.3(a) shows a chemical potential region for Li2MnO3with the boundaries set by the competing phases in the Li-Mn-O phase diagram.This chemical potential region represents the thermodynamically equilibrium conditions under which Li2MnO3could be stabilized.Here, to represent the chemical environment in Li-ion batteries, we focused on the Li-rich condition, as labeled in Fig.3(a).Fig.3(b) illustrates the calculated formation energies for a series of defects in Li2MnO3, including vacancies, antisites, and interstitials, as a function of the electron Fermi energy (EF).At the Li-rich condition, we found that theEFis pinned at 0.27 eV below the CBM by a pair of donor and acceptor defects (and), and at this,has the lowest formation energy of 0.56 eV among all the considered defects.These results indicated that, at the Li-rich condition, the defect formation in Li2MnO3should be difficult, due to the relatively large energies that are needed to create defects.Nevertheless, it is important to note that, as Li ions are extracted upon delithiation, theEFwould be continuously pushed down to the VBM.In this case, our calculations showed that various defects, such ascould form spontaneously in Li2MnO3, as they have negative formation energies.This result is in general consistent with the experimental observations that the cation migration and lattice oxygen loss usually occur in Li2MnO3upon delithiation[20,21].

Fig.3.(Color online) (a) The stable chemical potential region for Li2MnO3.(b) The formation energies of various defects in Li2MnO3, as a function of electron Fermi energy.(c) The schematic diagram for the charge transfer mechanism in Li2MnO3.
To get more insight into the defect formation in Li2MnO3upon delithiation, we proposed a defect charge transfer mechanism, as schematically illustrated in Fig.3(c).As discussed above, before delithiation, Li2MnO3is a semiconductor with its HOS (i.e., the VBM states) fully occupied.In this case, creating a donor defect in Li2MnO3would yield a defect level with occupied electrons, which usually leads to a large formation energy since it needs to break the strong Mn-O bonds, as shown in the left panel of Fig.3(c).As a consequence, before delithiation, defects cannot form easily in Li2MnO3.While, on the other hand, when Li ions are extracted from Li2MnO3upon delithiation, holes are injected into its HOS.Thus, the electrons originally occupied the defect levels can now drop into these hole states and gain an energy ( ΔE), as illustrated in the right panel of Fig.3(c).If ΔEis large enough to compensate the energy cost for breaking the Mn-O bonds, defects could form spontaneously.Given that the HOS of Li2MnO3are mainly determined by O 2porbitals and thus have a low energy, ΔEcould be large, which should be mainly responsible for the observed defect formation in Li2MnO3upon delithiation[20,21].
Compared to Li2MnO3, it has been widely observed that Li2RuO3could have a greatly improved cycling reversibility[22,23,25].To understand this phenomenon, we next discussed the electronic structure and defect properties of Li2RuO3.Fig.4(a) plots the spin-polarized band structure and PDOS for Li2RuO3.Different from its 3dcounterpart, our calculationsindicatedthatLi2RuO3isametalwithitsEFcrossing onlythespin-downchannel.Thishappensbecausethe large crystal field splitting of Ru 4dorbitals results in a lowspin magnetic ground state (see Table 1), as illustrated by the molecular-orbital diagram in Fig.4(b).Accordingly, the HOS of Li2RuO3should correspond to thestates that have a predominant Ru 4dcharacter.Thus, compared to Li2MnO3, we can expect that the HOS of Li2RuO3should have a much higher energy, which would prevent it from the defect formation, according to the defect charge transfer mechanism.Actually, by aligning the band structures of these two cathodes,we found that the HOS of Li2RuO3have an energy of 1.68 eV higher than that of Li2MnO3.Moreover, we also calculated the formation energies of several important defects in Li2RuO3at the Li-rich condition, including RuLi, Rui, Lii, and VO.The results indicated that all these considered defects have relatively large formation energies, with the smallest value of 3.33 eV found for VO.Notably, different from Li2MnO3, there are nearly infinite numbers of electrons and holes at the HOS of metallic Li2RuO3and thus the electrons originally occupying the defect level may jump into the HOS.For instance, the formation energy ofwas calculated to be 2.35 eV, which is smaller than that of, due to the charge transfer from the defect level to the HOS.It should be noted that the delithiation would not significantly change the energy of HOS and thus the defect properties of Li2RuO3.These calculations provide a good explanation on the improved cycling reversibility of Li2RuO3.
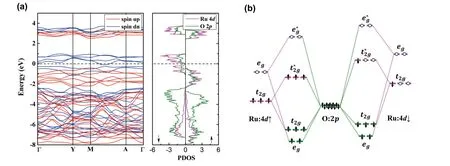
Fig.4.(Color online) (a) The band structure and projected density of states for Li2RuO3 and (b) the corresponding molecular-orbital diagram.
4.Conclusion
In this work, based on the first-principles calculations with the SCAN functional, we systematically studied the electronic structures and defect properties of two prototype Lirich TM oxides, Li2MnO3and Li2RuO3, in order to get more understanding on the structural degradation of these highcapacity cathodes upon delithiation.Our results indicated that the structural and cycling stability of Li2MnO3and Li2RuO3depend closely on their electronic structures, especially the energy of their HOS, from which the compensating electrons would be extracted upon delithiation.We found that Li2MnO3is a semiconductor with a band gap of 2.1 eV and thus its HOS correspond to the VBM states that are mainly determined by O 2porbitals.The defect calculations showed that, before delithiation, Li2MnO3is stable against the defect formation, due to the relatively large energies that are needed to create defects.Whereas, upon delithiation, we found that various defects could form spontaneously in Li2MnO3, as their formation energies become negative.The underlying mechanism of this phenomenon was ascribed to the charge transfer from defect levels into the injected holes at the HOS of Li2MnO3upon delithiation, which could gain an energy that is sufficiently large to compensate the energy cost for the defect formation.While, on the other hand, different from Li2MnO3, Li2RuO3was found to be a metal with its HOS predominately determined by Ru 4dorbitals and thus having a relatively high energy.As a consequence, the charge transfer from defect levels to hole states at its HOS could gain only a small energy that is not sufficient to trigger the defect formation, which explains why a good cycling reversibility could be realized in Li2RuO3.
Acknowledgements
This work was supported by Guangdong Basic and Applied Basic Research Foundation (2021A1515010219,2022A1515011990, and 2023A1515030086), Shenzhen High-Talent Research Foundation (827-000561), Shenzhen Science and Technology Program (KQTD20180412181422399 and JCYJ20220531102601004), High-Level University Construction Funds of SZU (866-000002110709), National Key R&D Program of China (2019YFB2204500), Science and Technology Innovation Commission of Shenzhen(JCYJ20180507181858539), and the National Natural Science Foundation of China (11991060, 12088101, and U2230402).In addition, we also gratefully acknowledge HZWTECH for providing computation facilities.
 Journal of Semiconductors2024年4期
Journal of Semiconductors2024年4期
- Journal of Semiconductors的其它文章
- Countermeasure against blinding attack for single-photon detectors in quantum key distribution
- Hybrid bonding of GaAs and Si wafers at low temperature by Ar plasma activation
- Improvement of Ga2O3 vertical Schottky barrier diode by constructing NiO/Ga2O3 heterojunction
- High-speed performance self-powered short wave ultraviolet radiation detectors based on κ(ε)-Ga2O3
- Effect of annealing on the electrical performance of N-polarity GaN Schottky barrier diodes
- On the relationship between imprint and reliability in Hf0.5Zr0.5O2 based ferroelectric random access memory