
叶下珠饮片质量标准优化研究*
2024-04-22 05:38胡冠宇谢嘉驰郭宇鸽
中医药导报 2024年3期
黎 旭,包 敏,胡冠宇,吴 茜,谢嘉驰,郭宇鸽
(1.湖南中医药大学第一附属医院,湖南 长沙 410007;2.株洲市食品药品检验所,湖南 株洲 412000;3.湘潭市中医医院,湖南 湘潭 411100)
叶下珠为大戟科叶下珠属植物叶下珠(Phyllanthus urinariaL.)的干燥全草,具有清热利湿、解毒消肿的功效,常用于痢疾、腹泻及传染性肝炎等疾病的治疗[1]。叶下珠含有木脂素[2]、香豆素[3]、萜类[4-5]、黄酮[6]、酚酸[7]、有机酸[8]及生物碱[9]等成分,具有抗病毒[10]、抗肿瘤[11]、抗菌[12]及保肝[13]等药理作用。
据本草考证[14],叶下珠作为药材最早收录于《生草药性备要》,有数百年药用史,但最早被收录的官方质量标准为1976年版《湖南省中药饮片炮制规范》[15]。目前,与其质量研究相关的文献报道有含量测定[16]、指纹图谱[17]、灰分、水分、醇溶性浸出物的测定[18-19]及生药鉴定[20]等。相比之下,2010年版《湖南省中药饮片炮制规范》存在标准缺项、质量控制方法单一等问题,不足以很好地控制湖南省市场上叶下珠饮片的质量。同时,为完善叶下珠饮片质量标准,湖南省市场监督管理局也启动了关于2010年版《湖南省中药饮片炮制规范》中叶下珠饮片标准修订的项目。基于此,本课题组开展了叶下珠质量标准研究,旨在为更好地监测湖南省市场叶下珠饮片的质量提供技术支撑。
1 材料
1.1 仪器 XSE 205 DV型电子天平(梅特勒-托利多仪器有限公司);2695高效液相色谱仪(沃特世科技有限公司);BX-61型电子显微镜(奥林巴斯有限公司);DHG-9147A型电热恒温干燥箱(上海精宏实验设备有限公司);SX-4-10型马弗炉(北京市永光明医疗仪器有限公司)。
1.2 药物与试剂 没食子酸(批号:110831-201906,纯度:91.5%)、柯里拉京(批号:111623-200301,纯度:100%)、叶下珠对照药材(批号:121374-201802)均购于中国食品药品检定研究院;色谱纯乙腈、甲醇、二氯甲烷、三氯甲烷和正己烷,均购于霍尼韦尔有限公司。
从不同生产企业收集叶下珠饮片共计11批,样品信息见表1。所有批次药材经株洲市食品药品检验所胡冠宇主管药师鉴定为大戟科叶下珠属植物叶下珠(Phyllanthus urinariaL.)的干燥全草。样本存放于株洲市食品药品检验所。
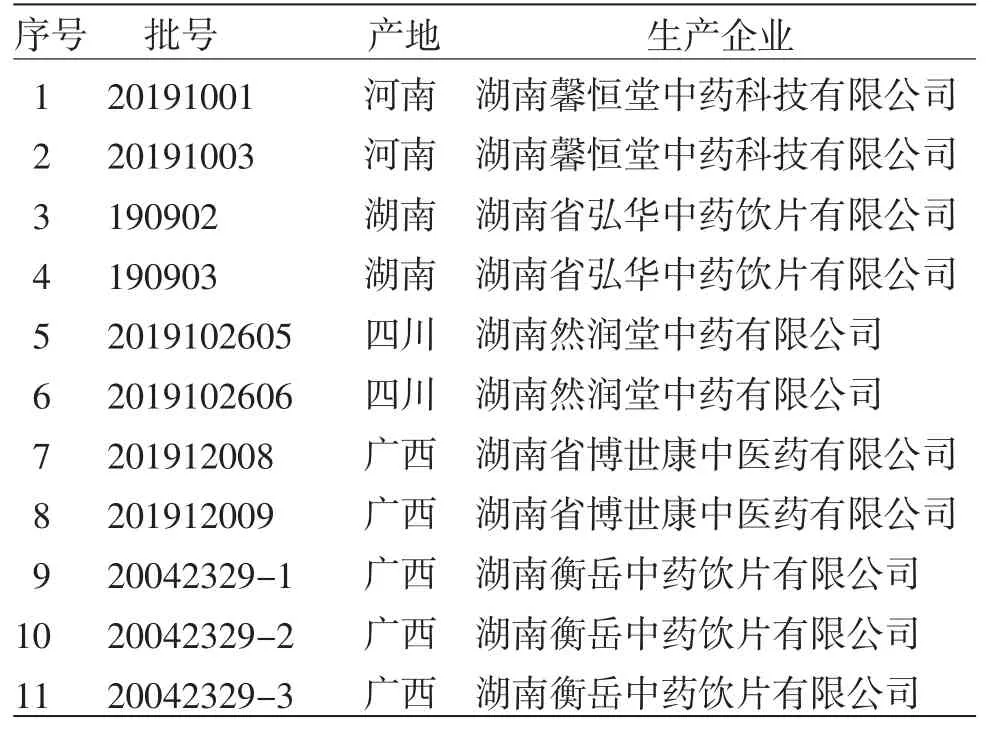
表1 叶下珠饮片信息
2 方法与结果
2.1 薄层鉴别 取本品粉末2 g,加乙醇20 mL,振摇2 h,滤过,滤液浓缩至2 mL,加硅胶(100~200目)2 g,拌匀,挥干溶剂,装入色谱柱中,以8 mL乙醚洗除杂质,弃去;加乙醇8 mL洗脱,收集乙醇洗脱液,蒸干;残渣加甲醇1 mL使溶解,制成供试品溶液;另取叶下珠对照药材2 g,同法制成对照药材溶液。按照2020年版《中华人民共和国药典》四部通则0502薄层色谱法,吸取上述两种溶液各10 μL,分别点样于同一硅胶G薄层板上,点样方式为手动点样,以三氯甲烷-甲醇(7.5:1)作为展开剂,上行展开;展开前,预饱和时间为30 min,环境温度为25.6 ℃,环境湿度为63%,展距约为10 cm;展开后,取出,晾干,喷以10%硫酸乙醇溶液,105 ℃加热至斑点显色清晰。于日光下视检。结果显示,在TLC色谱图中,供试品与叶下珠对照药材在相同位置上显相同颜色的斑点。(见图1)
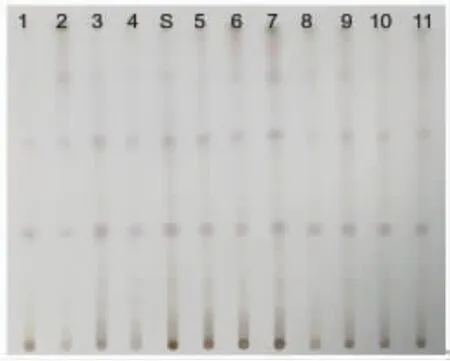
图1 TLC 图
2.2 粉末鉴别 取细粉(过5号筛)少许,置于载玻片,加1~2滴水合氯醛,置于酒精灯上透化至近无色,取下;加1滴甘油后装片,用吸水纸吸去多余甘油,置于显微镜下观察并拍照。通过对收集到的11批叶下珠饮片进行粉末显微鉴别,结果显示11批样品均能观察到叶表皮细胞、气孔、木栓细胞、韧皮纤维、木纤维、导管、非腺毛、草酸钙簇晶及果表皮细胞。具体显微鉴别特征为“本品粉末黄绿色。叶表皮细胞垂周壁波状弯曲,气孔多为平轴式,少数为不等式。木栓细胞呈类多角形或类方形,黄棕色,壁稍增厚,微弯曲。韧皮纤维成束或散在,长梭形,两端渐尖长。木纤维多成束,壁稍厚,木化,具单斜纹孔或短缝状纹孔。导管为螺纹、网纹,直径25~60 μm。非腺毛1~5个细胞,长40~210 μm,直径约30 μm,壁具角质纹理或壁疣。果皮表面细胞类圆形或多角形,有的角隅处增厚。草酸钙簇晶众多,直径10~20 μm,棱角较钝”。(见图2)
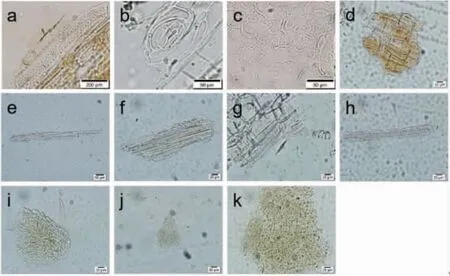
图2 粉末显微图
2.3 横切面鉴别 取叶下珠饮片,选择大小适中的茎数根、叶数片,置于装有水的培养皿中,浸泡2 h,软化,取茎或叶,用泡沫夹住,刀片进行切削,毛笔蘸取粘在刀片上的茎或叶横切薄片,置于水中伸展,然后用毛笔蘸取厚度合适的薄切片置于载玻片上,加清水1~2滴,盖上盖玻片,用吸水纸吸去多余清水,置于显微镜下观察。
(1)茎横切面鉴别:表皮细胞1列,细胞小,呈切向延长的长方形或长多角形,排列紧密,外被角质层。皮层由5~6层细胞组成;靠近表皮的1~2层细胞较小,切向延长,部分角隅处增厚形成厚角组织;其下1~2层细胞较大,排列不规则;靠近韧皮部的1~2层细胞呈类圆形或多角形。韧皮部较薄,由1~3层细胞组成;其外侧有中柱鞘纤维,成束,断续排列成环,壁较厚,层纹明显。形成层成环,不甚明显。木质部成环位于韧皮部内方;导管直径25~45 μm,呈类方形、近圆形或类多角形,径向单列或散在;木纤维排列紧密,成束或径向排列,壁较厚;射线由1~3列细胞组成。髓较大,约占整个断面的1/2,细胞较大,排列疏松,或部分中空。(见图3)
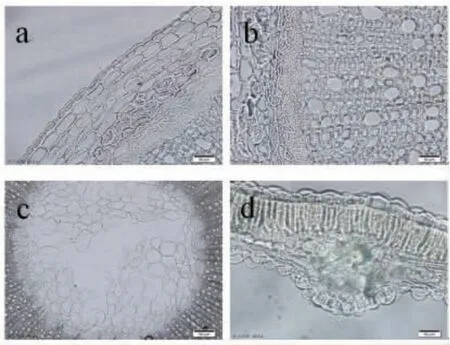
图3 茎和叶的横切面显微图
(2)叶横切面鉴别:上、下表皮均为1列细胞,且上、下表皮细胞常向外突起,外被角质层;细胞呈方形或类方形,排列紧密,垂周壁略弯曲,可见气孔。栅栏组织1列细胞,通过主脉;栅栏细胞呈长圆柱形,排列紧密,长45~70 μm,其中散有草酸钙簇晶。海绵组织由2~3列排列疏松的不规则的薄壁细胞组成,有少数小草酸钙簇晶散在。主脉在叶背面显著突起,外韧型;木质部导管4~5列径向排列,每列1~5个;韧皮部外侧有纤维散在,纤维壁厚,层纹明显;下表皮内的主脉维管束处有厚角组织,厚角细胞1~2列,细胞形状不规则,较大。(见图3)
2.4 含量测定
2.4.1 色谱条件 采用Shimadzu VP-SOD C18色谱柱(150 mm×4.6 mm,5 μm),以0.1%乙酸(A)-乙腈(B)为流动相,洗脱程序为0~7 min,5%B→10%B;7~30 min,10%B→15%B;30~34 min,15%B→17%B;34~36 min,17%B→80%B;36~37 min,80%B→5%B,36~37 min,5%B,流速为0.8 mL/min,采集时间为38 min,柱温为30 ℃,进样量为10 μL,紫外检测器,最佳吸收波长为280 nm。
2.4.2 溶液的制备
2.4.2.1 混合对照品溶液的制备 精密称取不同质量的没食子酸和柯里拉京对照品,置10 mL容量瓶中,加甲醇溶解并定容至刻度,摇匀,即得。经含量折算后,上述各对照品的质量浓度分别为996.0、1980 μg/mL。
2.4.2.2 供试品溶液的制备 取叶下珠饮片粉末(过3号筛)约1.0 g,精密称定,置具塞锥形瓶中,精密加入75%甲醇溶液40 mL,称定质量,超声(功率:100 W,频率:35 kHz)提取30 min,静置至室温,再称定质量,用75%甲醇溶液补足减失的质量,摇匀,滤过,取续滤液作为供试品溶液。
2.4.3 专属性试验 按照“2.4.1”项下色谱条件,分别进样10 μL空白、混合对照品溶液和供试品溶液进行专属性分析。结果表明供试品溶液在2种对照品保留时间(±0.5%)处都有特征峰,且分离度、拖尾因子及理论塔板数均满足定量分析要求,其他成分峰对待测成分无干扰。(见图4)
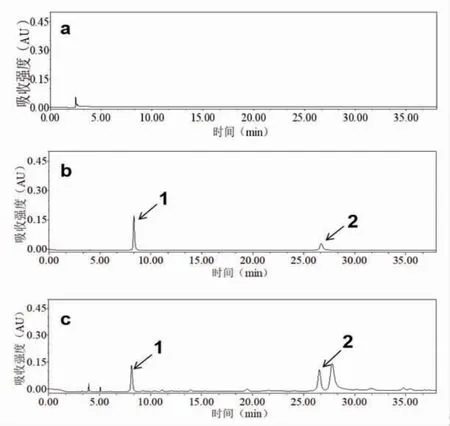
图4 HPLC 图
2.4.4 线性关系考察 精密量取混合对照品溶液适量,用75%甲醇稀释得系列质量浓度的混合对照品溶液(没食子酸质量浓度分别为12.45、24.90、49.80、124.50、249.00、498.00 μg/mL;柯里拉京质量浓度分别为24.75、49.50、99.00、198.00、495.00、990.00 μg/mL),按“2.4.1”项下色谱条件进行测定,以质量浓度X(μg/mL)为横坐标,峰面积Y为纵坐标,计算2个成分的线性回归方程。结果得没食子酸线性方程为Y=15 068X-14 509(r=0.999 8)、柯里拉京线性方程为Y=38 265X-7 032(r=0.999 9),表明没食子酸、柯里拉京分别在12.45~498.00、24.75~990.00μg/mL范围内线性关系良好。
2.4.5 精密度试验 取同一批样品(批号:190902),按照“2.4.2.2”项下方法制备供试品溶液,按照“2.4.1”项下色谱条件连续进样6次,记录各成分峰面积并计算相对标准偏差(RSD)。结果没食子酸、柯里拉京峰面积RSD值分别为0.62%、0.59%,表明仪器精密度良好。
2.4.6 稳定性试验 取同一批样品(批号:190902),按照“2.4.2.2”项下方法制备供试品溶液,分别于0、2、4、8、12 h按照“2.4.1”项下条件进样分析,记录各成分峰面积。结果没食子酸、柯里拉京峰面积RSD值分别为1.68%、1.75%,表明供试品溶液在12 h内稳定性良好。
2.4.7 重复性试验 取同一批样品(批号:190902),称取6份,按照“2.4.2.2”项下方法制备供试品溶液,按照“2.4.1”项下色谱条件进样分析,记录各成分峰面积。结果没食子酸、柯里拉京峰面积RSD值分别为1.04%、0.99%,表明该方法重复性良好。
2.4.8 加样回收率试验 取已知含量的样品(批号:190902)9份,每份约0.5 g,精密称定,分别加入绝对含量接近样品50%、100%、150%量的混合对照品溶液,每个浓度3份,按“2.4.2.2”项下方法分别制备供试溶液,按照“2.4.1”项下色谱条件进样分析,计算加样回收率。结果没食子酸、柯里拉京的平均加样回收率分别为100.14%、99.71%,RSD值分别为2.00%、2.02%,表明本方法准确度良好。(见表2)

表2 叶下珠饮片中没食子酸、柯里拉京加样回收率试验结果 (n=9)
2.4.9 样品含量测定 精密称取11批叶下珠饮片粉末各2份,分别按照“2.4.2.2”项下方法制备供试品溶液,按照“2.4.1”项下色谱条件进样测定,根据线性回归方程,计算没食子酸、柯里拉京的含量。(见表3)

表3 样品含量测定结果 (n=2)
2.5 水分、总灰分、酸不溶灰分、醇溶性浸出物测定 按照2020年版《中华人民共和国药典》四部通则0832第二法测定11批叶下珠饮片的水分;按照2020年版《中华人民共和国药典》四部通则2302测定11批叶下珠饮片的总灰分;按照2020年版《中华人民共和国药典》四部通则2302测定11批叶下珠饮片的酸不溶灰分。按照2020年版《中华人民共和国药典》四部通则2201测定11批叶下珠饮片的醇溶性浸出物。(见表4)
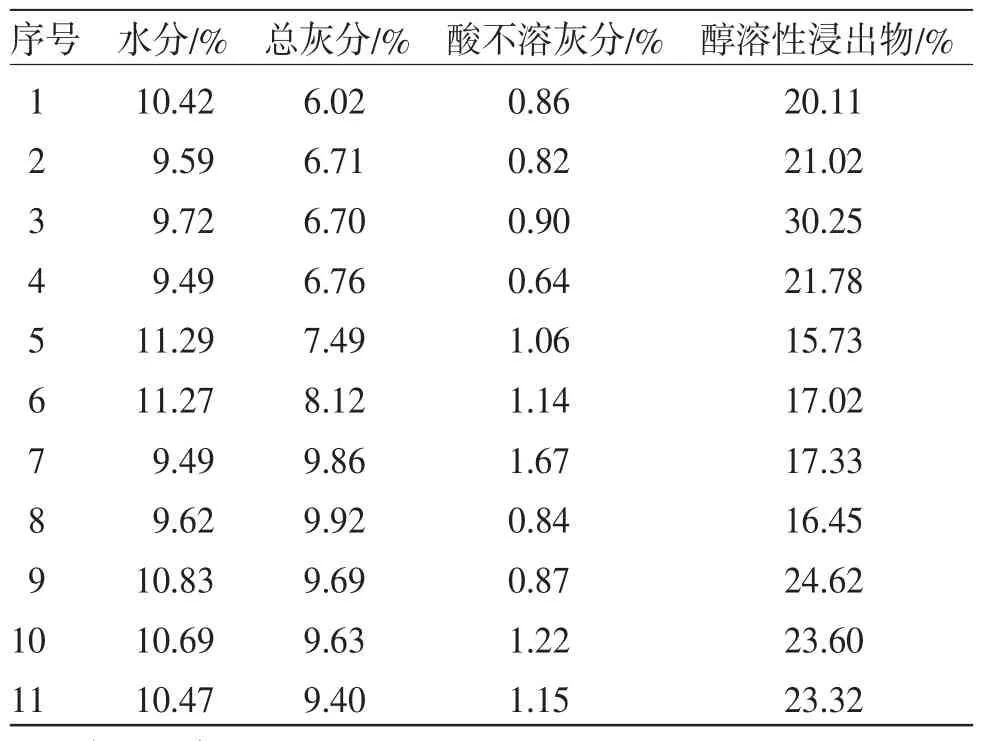
表4 叶下珠饮片中水分、总灰分、酸不溶灰分及醇溶性浸出物的含量 (n=3)
3 讨论
3.1 薄层色谱净化溶剂的选择 本研究比较了3种净化溶剂,含二氯甲烷、乙酸乙酯、乙醚。结果二氯甲烷杂质去除能力弱,背景显色深,目标点不清晰;乙酸乙酯可去除Rf值为0.6的目标点,影响鉴别结果;乙醚可选择性保留目标点,为最佳净化溶剂。(见图5)

图5 不同净化溶剂薄层色谱图
3.2 薄层色谱展开剂的选择 本研究比较了三氯甲烷-甲醇(7.5:1.0)、二氯甲烷-甲醇(6:1)和正己烷-乙酸乙酯-甲醇(6:2:1)3种展开剂。结果显示,与其他两种比较,三氯甲烷-甲醇(7.5:1.0)对Rf=0.4和Rf=0.7两紫红色斑点,展开效果均好,斑点清晰,为最佳展开剂。(见图6)
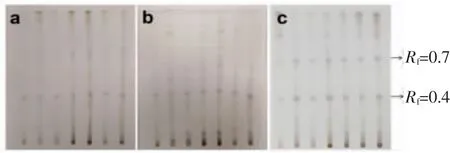
图6 不同展开剂薄层色谱图
3.3 显微鉴别 目前多个质量标准和文献记载了叶下珠横切面和粉末显微特征,与本研究大多相似或相同,但仍存在一定差异。如栅栏细胞长度,本研究为“45~70 μm”,湖北省中药材质量标准[21]为“13~17 μm”,孙佳石等[22]为“12~18 μm”。导管类型。本研究中导管为“螺纹、网纹”,张秀桥等[20]和湖北省中药材质量标准[21]为“多为螺纹、网纹、孔纹、少数环纹”,孙佳石等[22]为“多为螺纹、孔纹,少数环纹”,彭菲[23]等为“多为螺纹、孔纹,少数网纹导管”,干国平等[24]为“主为螺纹、网纹及孔纹,少数环纹”。气孔类型,本研究为“多为平轴式,少数为不等式”,彭菲等[23]、牛晓峰等[25]为“平轴式”,江西省中药饮片炮制规范[26]为“直轴式”。另有,《江西省中药饮片炮制规范》[26]中有观察到石细胞等组织,本研究均未发现。
3.4 含量测定
3.4.1 供试品溶液制备条件考察 本研究基于供试品溶液中没食子酸和柯里拉京峰面积最大或不再增加,比较了不同提取溶剂、提取时间及提取体积。结果显示,相对于50%甲醇和90%甲醇,75%甲醇所提取供试品溶液中没食子酸和柯里拉京峰面积最大;当提取时间为30 min,两种成分峰面积不再增加;当提取体积为40 mL时,峰面积亦不再增加。综上,选定40 mL 75%甲醇提取30 min为最佳提取条件。(见表5)
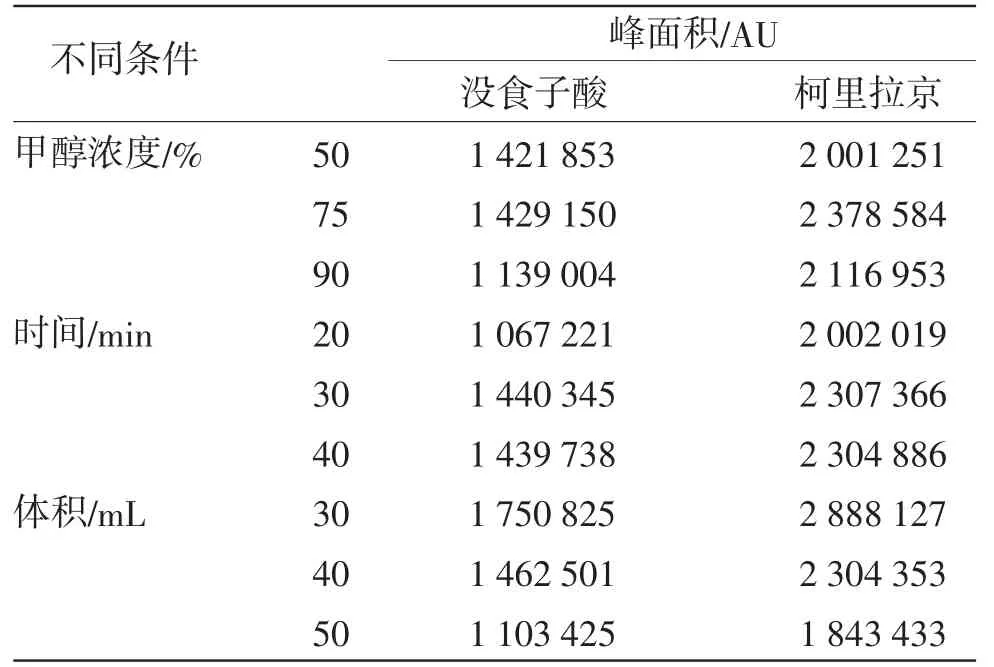
表5 不同考察条件下样品峰面积
3.4.2 色谱条件考察 本研究比较了不同流速(含0.8、1.0mL/min;不同流动相组成(乙腈-0.1%乙酸、乙腈-水)对分离效果的影响。结果当流速为1.0 mL/min时,柯里拉京和杂峰无法达到基线完全分离(见图7a);当流速为0.8 mL/min时,分离得到改善,但是峰出现明显前延和拖尾(见图7b);在流动相系统水相中加入0.1%乙酸后情况得到明显改善(见图7c)。

图7 不同流动相组成、流速色谱图
3.4.3 指标成分含量分析及限量值拟定 根据试验结果,本研究11批样品没食子酸含量为0.011 0%~0.258 2%,柯里拉京含量为0.1267%~0.3291%;已有文献报道,没食子酸为0.0391%~2.300 0%[27-28],柯里拉京为0.0341%~2.2140%[29-30];且韩越等[18]拟定“柯里拉京不得低于0.3%”,陕西省中药材标准[31]规定“柯里拉京含量不得低于0.1%”,同时尚未发现有标准将没食子酸纳入叶下珠含量指标成分。本研究综合研判,拟定“柯里拉京含量不得低于0.1%”,并新增没食子酸为含量指标成分,且拟定“没食子酸含量不得低于0.01%”。
3.5 水分、总灰分、酸不溶灰分、醇溶性浸出物含量分析和限量值拟定 根据试验结果,本研究11批样品水分含量为9.49%~11.29%,韩越等[18]为9.71%~10.75%,已发布标准[1,26,32]限量值分别为10%、12%、13%。总灰分为6.02%~9.92%,韩越等[18]为9.56%~14.44%,莫我跃等[19]为5.69%~8.02%,已发布标准[26,32]限量值分别为10%、15%。酸不溶灰分含量为0.64%~1.67%,莫我跃等[19]为0.74%~1.64%,已发布标准[26,32]限量值分别为3%、8%。醇溶性浸出物含量为15.73%~30.25%,韩越等[18]为16.16%~27.43%,莫我跃等[19]为16.35%~28.82%,已发布标准[32]限量值为10%。本研究综合研判,水分限度拟定为“不得过13.0%”,总灰分限度拟定为“不得过12.0%”,酸不溶灰分限度拟定为“不得过3.0%”,醇溶性浸出物限度拟定为“不得少于15.0%”。
综上所述,本研究在2010年版《湖南省中药饮片炮制规范》基础上,增加了粉末鉴别、茎横切面鉴别、叶横切面鉴别、水分、总灰分、酸不溶灰分、醇溶性浸出物及含量测定等,并优化了各项参数,且研究成果已经被2021年版《湖南省中药饮片炮制规范》收录,能够更为准确、全面地控制叶下珠饮片的质量。
猜你喜欢
选煤技术(2022年2期)2022-06-06
选煤技术(2022年2期)2022-06-06
世界科学技术-中医药现代化(2021年5期)2021-11-05
选煤技术(2021年6期)2021-04-19
四川蚕业(2021年4期)2021-03-08
中成药(2019年12期)2020-01-04
疯狂英语·新读写(2018年3期)2018-11-29
读者·校园版(2014年21期)2014-05-14
中国现代中药(2012年6期)2012-10-30
世界中医药(2010年3期)2010-08-02
