
以双上睑紫红斑为表现的周期性发热-阿弗他口炎-咽炎-淋巴结炎一例
2024-04-22 07:08刘雅芹罗志强谢映霞陈留鑫邓诗音蔡艳霞
中国麻风皮肤病杂志 2024年5期
刘雅芹 罗志强 谢映霞 陈留鑫 邓诗音 蔡艳霞
1 广东医科大学附属医院皮肤科,广东湛江,524000;2 广东医科大学,广东湛江,524000
周期性发热-阿弗他口炎-咽炎-淋巴结炎(periodic fever,aphthous stomatitis,pharyngitis and adenitis,PFAPA)综合征是儿童时期最常见的自身炎症性疾病(AUIDs),由Marshall等[1]在1987年首次提出。典型PFAPA的临床表现主要为周期性发热伴阿弗他口炎、咽炎及颈部淋巴结肿大中至少1种[2]。其中扁桃体炎最常见,阿弗他口炎最少见。皮疹是其最常见的非特异性表现[3],然而既往多数报道都缺乏对皮疹形态的详细描述。我们报道1例以反复口腔溃疡合并全身散在水肿性紫红斑为主要表现的PFAPA患儿,并就相关文献进行复习,以增加皮肤科医生对该病的诊治,避免误诊。
临床资料患儿,男,10岁。因“反复口腔溃疡10年,加重伴发热6个月余。”于2023年08月至我科就诊。患儿自6月龄开始反复口腔多发溃疡,间隔1~2个月发作1次,每次持续约1周愈合,愈后不留瘢痕,无发热、关节痛,无结膜、生殖器溃疡等,至当地诊所诊治效果欠佳,病情反复。6个月余前于公园游玩后再次出现口腔溃疡,数量较前增多,疼痛明显,伴发热,热峰38.5℃,同时出现咽痛,四肢红斑、结节,伴疼痛、瘙痒,左眼结膜红肿,双踝关节肿痛,双大腿肌肉疼痛、乏力,行走困难,至外院就诊查血清总IgE大于2500 IU/mL、CRP升高36.28 mg/L,考虑“过敏性皮炎”,予退热、抗组胺、抗感染治疗后好转。此后上述症状反复出现,呈周期性发作,约2周1次,5~7天好转,体温波动在38.0℃~38.5℃,多次至当地医院诊治,予“西替利嗪、地塞米松、克林霉素、丙种球蛋白”等治疗后症状可好转。4天前上述症状再发伴双侧眼睑水肿性紫红斑。病程中无咳嗽、咳痰、流涕等呼吸道症状,发热间期无任何不适,生长发育同正常儿童。既往史及出生史无特殊。家族中父亲、奶奶、叔叔、姑姑有反复口腔溃疡病史,但无反复发热,母亲及弟弟体健。
系统查体:咽轻度充血红肿,扁桃体未见肿大;心肺腹无异常,全身浅表淋巴结无肿大,四肢肌力正常。皮肤科查体:唇、颊黏膜、舌底可见7个溃疡,圆形或类圆形,直径0.2~0.5 cm,中央有黄白色基底,周围红晕,触痛(图1a);双上眼睑见水肿性紫红斑(图1b);四肢及背部见散在分布紫红色斑、结节,色素沉着斑(图1c),双肘部伸侧紫红色斑疹(图1d、1e)。
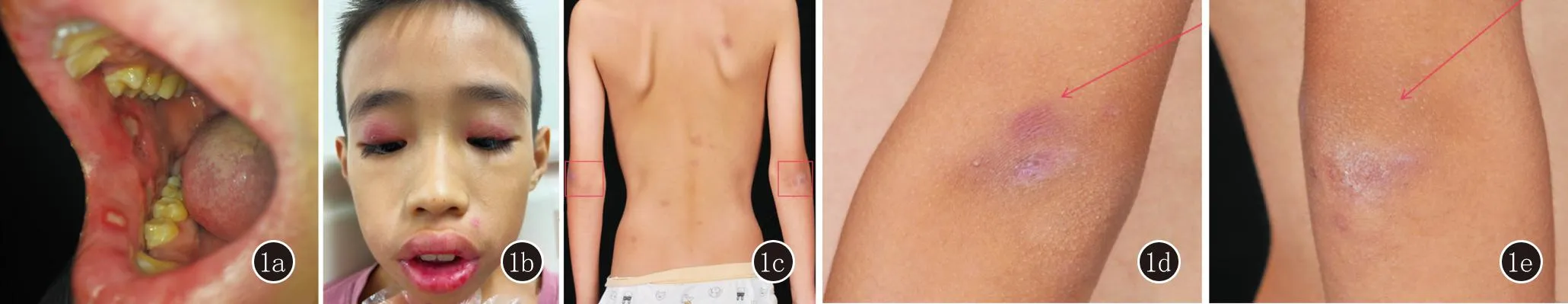
图1 1a:口唇多发溃疡;1b:双上眼睑紫红色水肿性红斑;1c:四肢及背部见散在紫红色斑、结节,色素沉着斑;1d、1e:双肘部伸侧紫红色斑疹
实验室检查:白细胞4.84×109/L[(4.0~10.0)×109/L],中性粒细胞比例30.50%↓(50%~70%),中性粒细胞绝对值1.48×109/L ↓[(2.00~7.00)×109/L],淋巴细胞比例58.10%↑(20.00%~40.00%),血红蛋白109.00 g/L↓(120~160 g/L)。免疫球蛋白总IgE 2510 IU/mL↑(0.00~165.30 IU/mL)。尿常规、大便常规、肝肾功能、凝血四项、ESR、CRP、PCT未见异常。风湿四项、抗核抗体系列、抗中性粒细胞浆抗体3项、磷脂综合征3项、肌炎抗体谱均阴性。巨细胞病毒抗体IgM、风疹病毒抗体IgM、EB病毒抗体IgM、肺炎支原体抗体、HHV6、单纯疱疹II型抗体IgM、弓形体抗体IgM均阴性。寄生虫抗体:肝吸虫IgG抗体(+)。肥达试验+外斐试验阴性。胸部DR:双肺纹理稍增粗;左膈面胸膜局部增厚粘连。心脏、肝胆胰脾及泌尿系彩超未见异常;双侧大腿肌电图未见异常。
口腔皮损组织病理示:表皮不规则增生,棘层海绵水肿,真皮浅层水肿,血管周围淋巴细胞及中性粒细胞浸润,肌肉间淋巴细胞浸润(图2)。

图2 表皮不规则增生,棘层海绵水肿,真皮浅层水肿,血管周围淋巴细胞及中性粒细胞浸润,肌肉间淋巴细胞浸润(2a:HE,×40;2b:HE,×100)
诊断:周期性发热-阿弗他口炎-咽炎-淋巴结炎。
患儿入院后已无发热、关节疼痛,遂予地塞米松磷酸钠+碳酸氢钠+0.9%氯化钠配制溶液漱口,人表皮生长因子外用溶液外喷,口服头孢呋辛酯片及西替利嗪滴剂对症治疗。1周后双眼睑红斑消退,口腔溃疡好转。出院后2个月电话随访,间中发作1次,至诊所予地塞米松治疗后好转,目前口腔溃疡尚未痊愈,无发热、咽痛、肌痛、皮疹等。
讨论PFAPA是一种自限性的发热综合征,常于5岁前起病,近年来也有不少报道于儿童期及成年发病患者。据挪威统计5岁以下儿童的发病率为0.23‰,男童发病率略高于女童[4]。PFAPA病因及发病机制尚不明确,目前认为由多基因变异、遗传易感性、以及环境因素等共同导致[5,6]。免疫失调可能是本病潜在基本机制,近期研究表明,在PFAPA发作期间先天性免疫系统的不同成分失调,可能被环境刺激(例如微生物等)激活,提示炎症小体介导的先天免疫系统激活可能是PFAPA综合征的第一步,随后效应T细胞活化再分布可能解释了该疾病的周期性发作[5,7]。
PFAPA特征性临床表现为发热、阿弗他口炎、咽炎和颈部淋巴结肿大周期性发作[2]。除发热外,欧洲患儿最常见的是红斑性或渗出性咽炎,中国儿童最常见的是反复发作的扁桃体炎,阿弗他口炎最少见[7]。患儿发作期还可出现一些非特异性表现,包括皮疹、关节痛、腹痛、结膜炎、眶周水肿、腹泻、肌痛及头痛等,其中皮疹最常见[3]。由于患儿多就诊于儿科或耳鼻喉科,既往多数报道都缺乏对皮疹形态的详细描述。有报道称以红斑为主,多见于躯干、掌足底斑疹或紫癜[8]。
目前PFAPA尚未有统一的诊断标准,Takeuchi等[9]通过对199例患儿的临床表现实验室指标的变化及治疗反应进行综合分析,基于1987年Marshall等[1]提出、1999年Thomas等[10]修正的标准,提出来Takeuchi标准如下:(1)必须标准:体温>38℃ 以上,持续时间<8 d(平均4 d),至少重复4次。(2)支持标准:①发病年龄<5岁;②扁桃体炎或咽炎伴白苔;③无咳嗽;④伴有口疮性口炎、淋巴结炎、咽痛、呕吐、严重头疼等临床症状的任意一种;⑤家族史(包括反复发热、扁桃体炎或扁桃体切除病史);⑥相关炎症指标的实验室检查如CRP和SAA,在发作时异常升高,但在发作间期水平正常;⑦血清IgD水平升高;⑧糖皮质激素治疗敏感(半天内口服泼尼松0.5 mg/kg,发热可缓解)。确诊PFAPA:具备必须标准和至少5项支持标准;疑似PFAPA:具备必须标准和4项支持标准。由于目前已有年长儿,甚至成人PFAPA的报道,年龄限制不作为诊断的必备条件[10]。
本例患儿以反复口腔溃疡为首发表现,后出现发热,伴四肢红斑、结节,双大腿疼痛无力,随后双上眼睑出现水肿性紫红斑,根据临床表现极易误诊为Behcet综合征(Behcet syndrome,BS)、幼年皮肌炎(juvenile dermatomyositis,JDM)或重叠综合征等其他结缔组织病。BS除复发性口腔溃疡外同时还伴有复发性生殖器溃疡、眼部病变(以葡萄膜炎多见),皮肤病变常见结节性红斑、假性毛囊炎,无周期性发热;组织病理表现为白细胞破碎性闭塞性血管周围炎和静脉血栓形成,伴淋巴细胞浸润各种大小的毛细血管、静脉和动脉,可通过临床和组织病理鉴别[11]。JDM肌无力呈进行性加重而非周期性,典型皮肤表现除眶周水肿性紫红斑,还包括Gottron’s丘疹、Gottron’s征和甲襞毛细血管异常等,严重者出现吞咽困难,实验室检查抗Jo-1抗体阳性,血清肌酸激酶、乳酸脱氢酶、谷丙转氨酶或谷草转氨酶升高,可通过实验室检查及典型临床表现排除[12]。本例患儿血肌酶谱均正常,应注意无肌病性皮肌炎(amyopathic dermatomyositis,ADM)可能,ADM的诊断标准示具有特征性皮肌炎的皮疹超过6个月[13],而该患儿皮疹持续约10天自行消退,因此可排除。本例患儿临床表现为顽固性口腔溃疡、周期性发热(>38℃,1次/2周,持续5~7天),发作时伴咽痛、皮疹、关节痛、肌痛等症状,且发热期CRP升高,糖皮质激素治疗敏感,无咳嗽等上感症状,满足PFAPA诊断标准,最终确诊为PFAPA综合征。
值得一提的是,本例患儿自幼反复口腔溃疡,10岁时才出现周期性发热等症状。根据一项纳入2个欧美人群队列和1个土耳其人群队列研究表明,PFAPA综合征、复发性阿弗他口炎和BS发病机制具有共同特征,均与IL12A、IL10、STAT4 和CCR1-3 基因变异密切相关[6]。提议将三者划分为一个疾病大类,统称Behcet谱系障碍(Behcet spectrum disorders,BSD),病情从轻到重依次为复发性阿弗他口炎、PFAPA综合征和BS[6]。因此推测本例患儿早期的顽固性口腔溃疡为复发性阿弗他口炎,由于长期未规律诊治使过敏刺激激活体内炎症因子引起免疫失调而进展为PFAPA综合征。已有报道许多成人BS患者儿童早期的症状符合PFAPA的诊断标准[14]。本例患儿未来是否会进一步发展为BS,仍需要密切随访观察。
由于PFAPA具有自限性的特点,目前尚无统一标准的治疗方案,治疗的主要目标是控制急性发作、减轻临床症状及降低发作频率。首选糖皮质激素快速控制发热,西咪替丁和秋水仙碱作可延长发作间期,药物治疗无效或发热发作严重干扰生活的患者,可考虑行扁桃体切除术[15,16]。由于本例患者入院后已退热,遂仅予对症治疗,既往在外院多次使用地塞米松疗效可,这与文献报道相符。
综上,本文报道了1例自幼患复发性阿弗他口炎,后逐渐出现发热、水肿性紫红斑及关节痛、肌痛的PFAPA综合征患儿。该病首诊于皮肤科较少,临床上极易误诊,提示我们在临床上遇到此类患者,应详细询问病史,警惕PFAPA综合征。
猜你喜欢
今日农业(2021年21期)2021-11-26
少年文艺(2020年5期)2020-05-11
百科探秘·航空航天(2018年11期)2018-11-29
留学生(2017年4期)2017-06-16
中华老年口腔医学杂志(2016年3期)2017-01-15
中国医疗美容(2015年1期)2015-07-12
西南军医(2015年3期)2015-04-23
中国卫生标准管理(2015年8期)2015-01-26
中国中医药现代远程教育(2014年20期)2014-03-01
当代畜禽养殖业(2014年7期)2014-02-27
