
A comprehensive review of genetic causes of obesity
2024-04-14 10:06:36MarcioJosConcepciZavaletaJuanEduardoQuirozAldaveMardelCarmenDurandsquezElmanRolandoGamarraOsorioJuandelCarmenValenciadelaCruzClaudiaMercedesBarruetoCallirgosSusanLucianaPuellesLeElenadeJesAlvaradoLeFransLeivaCabrera
World Journal of Pediatrics 2024年1期
Marcio José Concepción-Zavaleta ·Juan Eduardo Quiroz-Aldave ·María del Carmen Durand-Vásquez ·Elman Rolando Gamarra-Osorio ·Juan del Carmen Valencia de la Cruz ·Claudia Mercedes Barrueto-Callirgos ·Susan Luciana Puelles-León ·Elena de Jesús Alvarado-León ·Frans Leiva-Cabrera ·Francisca Elena Zavaleta-Gutiérrez ·Luis Alberto Concepción-Urteaga ·José Paz-Ibarra
Abstract Background Obesity is a multifactorial chronic disease with a high, increasing worldwide prevalence.Genetic causes account for 7% of the cases in children with extreme obesity.Data sources This narrative review was conducted by searching for papers published in the PubMed/MEDLINE, Embase and SciELO databases and included 161 articles.The search used the following search terms: “obesity”, “obesity and genetics”,“leptin”, “Prader-Willi syndrome”, and “melanocortins”.The types of studies included were systematic reviews, clinical trials, prospective cohort studies, cross-sectional and prospective studies, narrative reviews, and case reports.Results The leptin-melanocortin pathway is primarily responsible for the regulation of appetite and body weight.However,several important aspects of the pathophysiology of obesity remain unknown.Genetic causes of obesity can be grouped into syndromic, monogenic, and polygenic causes and should be assessed in children with extreme obesity before the age of 5 years, hyperphagia, or a family history of extreme obesity.A microarray study, an analysis of the melanocortin type 4 receptor gene mutations and leptin levels should be performed for this purpose.There are three therapeutic levels: lifestyle modifications, pharmacological treatment, and bariatric surgery.Conclusions Genetic study technologies are in constant development; however, we are still far from having a personalized approach to genetic causes of obesity.A significant proportion of the affected individuals are associated with genetic causes;however, there are still barriers to its approach, as it continues to be underdiagnosed.
Keywords Leptin·Melanocortin·Obesity·Prader-Willi syndrome·Precision medicine
Introduction
Obesity is a chronic multifactorial disease characterized by the excessive accumulation of body fat [1, 2], which increases the risk of cardiovascular disease, diabetes,obstructive sleep apnea, dyslipidemia, non-alcoholic fatty liver disease, cancer, and mental health problems [3–5].
The prevalence of overweight is 26.5%, and that of obesity is 12.5%, both of which have increased in recent decades [4].In the Americas, the prevalence of obesity is 22.4%, with higher rates in the United States (23.3%) and Mexico (18.4%) [4].It mainly affects middle-aged adults in low-income countries and adults of all ages in high-income countries [3, 4].In the United States, the prevalence of obesity among children aged 2–5 years is 12.7%, between 6 and 11 years is 20.7%, and between 12 and 19 years is 22.2% [3, 6–8].In Southeast Asia, the prevalence of obesity in children and adolescents is between 3% and 5%; in the Western Pacific region, it ranges from 9% to 19%; in the Eastern Mediterranean region (North Africa and the Middle East), it is 11%; and in sub-Saharan Africa, it varies from 3% to 5% [9].On the other hand, overweight/obesity affects 60% of adults and nearly one in three children (29%of boys and 27% of girls) in the World Health Organization European Region [10].Additionally, in South America, the prevalence of obesity among children under 5 years of age is 9%, and for those over 5 years of age, it rises to 12% [11].
Modifiable environmental factors associated with obesity include excessive hypercaloric intake from sugar-sweetened beverages and fast food, as well as reduced energy expenditure due to automobile transportation and overuse of electronic devices [2, 7].Lifestyle has the capacity to induce reversible changes in gene expression without affecting the DNA sequence, a phenomenon known as epigenetics [12].Several genes implicated in obesity are epigenetically regulated [13].Additionally, socioeconomic status, overweight in caregivers,birth weight, environmental pollution, infections, and stress all play a role in the development of obesity [2, 14].
The theory of an endogenous cause of obesity was first proposed by Von Noorden in 1907, and since then, it has been widely investigated [15].Genetic causes associated with obesity account for 7% of cases of extreme obesity in children [16].Inheritability ranges from 40% to 70%, with cultural transmission playing a minor role [17].There are several gaps in the study of the genetic causes of obesity,such as lack of awareness, insufficient information on its pathophysiology, and limited access to specific genetic tests and effective treatments.Therefore, further research is necessary to improve our understanding of the genetic syndromes associated with obesity and to develop effective prevention and treatment strategies.
The goal of this review is to address the identified genetic causes of obesity and summarize the current literature about its physiopathology, classification, diagnosis, treatment, and precision medicine.
Pathophysiology
The leptin-melanocortin pathway (Fig.1) plays a crucial role in regulating appetite and body weight.Leptin, a hormone produced in adipose tissue, binds to its main receptors located in the arcuate nucleus of the hypothalamus.This binding increases the production of proopiomelanocortin (POMC),which is then processed by proprotein convertase subtilisin and kexin types 1 and 2 (PCSK1 and PCSK2).This processing results in the production of adrenocorticotropic hormone(ACTH) and melanocyte-stimulating hormone (MSH) [17].These two hormones are collectively referred to as melanocortins.MSH acts centrally on the melanocortin type 4 receptor (MC4R), which is highly expressed in the paraventricular nucleus of the hypothalamus.The activation of MC4R leads to reduced food intake and increased energy expenditure [17].Brain-derived neurotrophic factor (BDNF) plays a significant role in energy homeostasis [18].It originates in the hippocampus, is abundant in the hypothalamus and adipose tissue, and acts on both MC4R and tropomyosin-related kinase B receptor (TrkB) to decrease food intake [19–22].

Fig.1 Leptin-melanocortin pathway.ACTH adrenocorticotropic hormone, ArcN arcuate nucleus, BDNF brain-derived neurotrophic factor,LEPR leptin receptor, MC4R melanocortin 4 receptor, MSH melanocyte-stimulating hormone, PCSK1 proprotein convertase subtilisin/kexin type 1, PCSK2 proprotein convertase subtilisin/kexin type 2,POMC proopiomelanocortin, PVN paraventricular nucleus
Oxytocin, produced by the hypothalamus in its paraventricular and supraoptic nuclei, acts centrally, activating brain areas that exert cognitive control over eating and increasing the activity of areas related to food reward,decreasing food intake [23].In contrast, melanin-concentrating hormone originates in the lateral hypothalamus and acts centrally to increase appetite [24].
In addition to these hypothalamic pathways, other neural and gut hormonal pathways are involved in appetite regulation (Fig.2).Within the arcuate nucleus of the hypothalamus, there are appetite suppressor neurons (producing POMC) and appetite stimulatory neurons [producing neuropeptide Y (NPY) and agouti-related peptide (AgRP)][25].Peripheral hormones derived from the gut and adipose tissue act centrally in the hypothalamus, affecting energy intake and expenditure [26].Leptin stimulates POMC neurons and inhibits NPY/AgRP neurons, while insulin acts centrally, reducing intake by inhibiting NPY/AgRP neurons [25].Gut hormones cholecystokinin, peptide YY,pancreatic polypeptide, glucagon-like peptide 1 (GLP-1),nesfatin 1, oxyntomodulin, and uroguanylin mediate satiety signals in the paraventricular nucleus of the hypothalamus in addition to the stimulation of insulin production [26–28].Ghrelin is the only gut hormone that stimulates appetite through growth hormone-releasing hormone receptors on NPY neurons [29, 30].Endocannabinoids regulate intake by activating reward pathways within the brain [31].

Fig.2 Other neural and gut pathways involved in appetite regulation.AgRP agouti-related peptide, CCK cholecystokinin, GLP-1 glucagon-like peptide 1, NF-1 nesfatin 1, NPY neuropeptide Y, OXM oxyntomodulin, POMC proopiomelanocortin, PP pancreatic peptide, PYY peptide YY,UGN uroguanylin
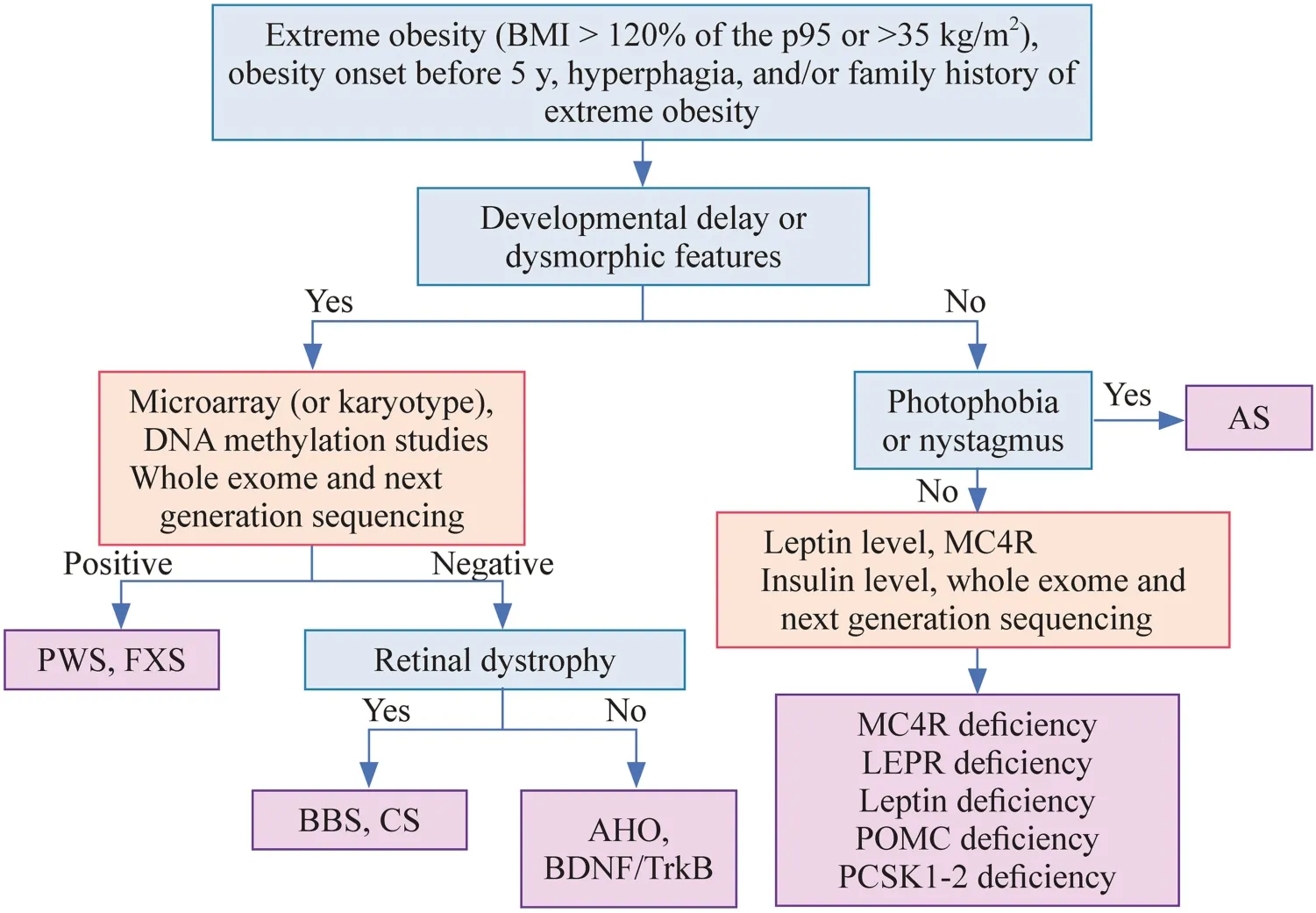
Fig.3 Diagnostic algorithm for the genetic-related causes of obesity.AHO Albright hereditary osteodystrophy, AS Alström syndrome,BDNF/TrkB brain-derived neurotrophic factor/tropomyosin-related kinase B receptor, BMI body mass index, BBS Bardet-Biedl syndrome,CS Cohen syndrome, DNA deoxyribonucleic acid, FXS fragile X syndrome, LEPR leptin receptor, MC4R melanocortin 4 receptor, PCSK1-2 proprotein convertase subtilisin/kexin types 1 and 2, POMC proopiomelanocortin, PWS Prader-Willi syndrome
The intestine has an important role in energy metabolism and is the habitat of a large resident microbiota, which includes species ofBacteroidetes,Firmicutes,Actinobacteria,Proteobacteria, andVerrucomicrobiain a delicate balance [27, 32].Obesity may be associated with an increase inFirmicutesand a decrease inBacteroidetes[33, 34].The alterations in the gut microbiota could impair the release of the previously described hormones, although these mechanisms have not yet been fully elucidated [27, 35–37].
Classification of genetic causes associated with obesity
There are three subtypes of the genetic causes of obesity:syndromic Mendelian obesity, non-syndromic (monogenic)Mendelian obesity, and polygenic obesity [2, 38].The first subtype is syndromic Mendelian obesity, which is caused by chromosomal abnormalities and rare variants of genes that encode crucial proteins involved in energy balance regulation and whose inheritance follows a Mendelian pattern,either autosomal or X-linked [39].These are rare pleiotropic syndromes involving reduced intellectual ability, dysmorphic features, and specific congenital alterations [39].Nonsyndromic causes, also known as monogenic obesity, manifest phenotypically when two dysfunctional copies of the genes are present in a homozygous or compound heterozygous manner, are rare and are characterized by increased intake and obesity [38, 40].Finally, polygenic obesity arises from the cumulative effects of multiple common genetic variants, does not have a clear inheritance pattern and is the most common form of obesity [41, 42].
There are several syndromes that have been described to date, some of which are yet to be named, while others have multiple names, contributing to confusion.The correct nomenclature for the newly identified genetic syndromes associated with obesity remains a subject of controversy [2].
Syndromic obesity
The most common cause of syndromic obesity is Prader-Willi syndrome (PWS), which affects 1 in 21,000 individuals, and it remains the primary cause of rapid deterioration and mortality in these patients [43].PWS is characterized by severe neonatal hypotonia and age-related feeding abnormalities, including anorexia and failure to thrive in infancy, followed by severe hyperphagia, compulsivity, and central obesity between 4 and 8 years of age, which appear to be the result of changes in intestinal hormones [38].Ghrelin levels are usually suppressed by food intake, but this does not happen in patients with PWS, all of whom have elevated ghrelin levels, occurring prior to the onset of heightened appetite.Most likely, the rise in ghrelin levels occurs early in infancy as a response to poor feeding, and persistent hyperghrelinemia promotes hyperphagia and obesity later in childhood [44].Patients with PWS also present an increase in resistin and adiponectin (lipogenic hormones) and a reduction in pancreatic polypeptide (anorexigenic hormone).No significant differences were found in the levels of leptin, obestatin, and peptide YY [44, 45].
Moreover, PWS may include facial dysmorphism, mild intellectual disability, behavioral abnormalities, delayed motor development, and hormonal deficiencies secondary to hypothalamic dysfunction.The latter includes central adrenal insufficiency, central hypothyroidism, short stature due to growth hormone deficiency, and hypoplasia of the genitals resulting from hypogonadotropic hypogonadism [43,45–48].There are several genes involved in PWS, located at chromosomal position 15q11.2.These genes includeMKRN3(makorin ring finger protein 3),MAGEL2(MAGE family member L2),NDN(necdin),NPAP1(nuclear pore associated protein 1),SNURF-SNRPN(SNRPN upstream reading frame-small nuclear ribosomal protein 1), andSNORD 116(small nucleolar RNA, C/D box 116 cluster)[49–51].The latter gene plays a crucial role in the regulation of food intake, and its microdeletion is associated with the development of the PWS phenotype [52].
Bardet-Biedel syndrome (BBS) is a rare autosomal recessive ciliopathy characterized by retinal dystrophy (94%),central obesity (72%–89%), post-axial polydactyly (79%),learning difficulties (66%), hypogonadism and genitourinary disorders (59%), renal dysfunction (52%), and other cardiovascular, neurological, gastrointestinal, and metabolic disorders [53, 54].Its prevalence varies depending on the consanguinity of the population [38, 55].At birth,post-axial polydactyly is the only manifestation, and other features slowly appear in the first decade of life [54].The gradual onset of night blindness, photophobia, and loss of central and/or color vision, which appear at 8.5 years of age,usually lead to a definitive diagnosis [38, 55, 56].Threequarters of individuals with BBS present obesity, although birth weight is usually normal.The prevalence of type 2 diabetes, hypogonadism, cognitive deficits, behavioral lability,speech deficits, and renal and cardiac abnormalities is high[54].To date, more than 20 mutations in genes associated with BBS have been described [57].
Alström syndrome (AS) shares clinical features with BBS, such as obesity and abnormalities related to vision,renal function, gonads, and height.However, the onset of visual problems in AS occurs earlier than in BBS, and unlike BBS, AS is not characterized by polydactyly and cognitive deficits [53, 58].Additionally, individuals with AS may also present with hyperinsulinemia, type 2 diabetes mellitus, dyslipidemia, and hepatic steatosis [59, 60].
Cohen syndrome is characterized by developmental delay,cognitive and behavioral disturbances, and typical facies such as downward slanting palpebral fissures, mild maxillary hypoplasia, prominent nasal root, micrognathia (with an open mouth expression that shows incisor prominence),myopia, and severe retinal dystrophy [61, 62].Infants with Cohen syndrome often have low birth weight and failure to thrive due to feeding difficulties.Later in childhood, they may develop central obesity.Short stature, pubertal delay,testosterone deficiency, hypogonadotropic hypogonadism,and neutropenia are also commonly observed [61, 63].
Albright hereditary osteodystrophy (AHO) is characterized by a rounded face, short stature, stocky body, brachydactyly, subcutaneous ossification, dental anomalies,delayed psychomotor development, and early-onset obesity and may include hypogonadism, other skeletal abnormalities, and cataracts [64].Conversely, it can present as pseudohypoparathyroidism (PHP), characterized by hypocalcemia,hyperphosphatemia, and elevated parathyroid hormone [65].If AHO presents without the hormonal alterations of PHP, it is known as pseudopseudohypoparathyroidism [64].
Fragile X syndrome (FXS), a common cause of mental retardation, is caused by an expansion of nucleotide repeats that leads to silencing of theFMR1gene [66, 67].Its most common features include a long face, large ears, joint hypermobility, mitral valve prolapse, and macroorchidism [68].At birth, infants may have hypotonia, poor sucking, and frequent regurgitation, and later, they may exhibit delayed psychomotor development, behavioral issues (such as autism spectrum disorders and compulsive disorders such as hyperphagia and aggressiveness), and 30%–60% may develop obesity [68].About 10% of individuals with FXS have severe obesity [69].Males are typically more severely affected due to having a single X chromosome [68].
Rapid onset obesity with hypothalamic dysfunction,hypoventilation, autonomic dysregulation, and neural tumor (ROHHADNET) syndrome is a rare cause of syndromic obesity [70].Approximately 40% of ROHHAD patients present with ganglioneuroma or ganglioneuroblastoma [71].The etiology of the syndrome has been postulated to involve epigenetic and autoimmune changes.However, candidate genes associated with neuronal development (BDNF and TrkB) or the hypothalamic and autonomic dysfunction pathway (5-hydroxytryptamine receptor 1A, orthopedia, pituitary adenylate cyclase activating polypeptide, hypocretin, hypocretin-receptor 1, hypocretin-receptor 2) did not show any significant genetic variants [72].Despite ongoing researchefforts, the exact cause of this syndrome remains unclear,and no significant genetic findings have been reported to date.The main causes of syndromic obesity are described in Table 1.Less frequent causes are summarized in Table 2.
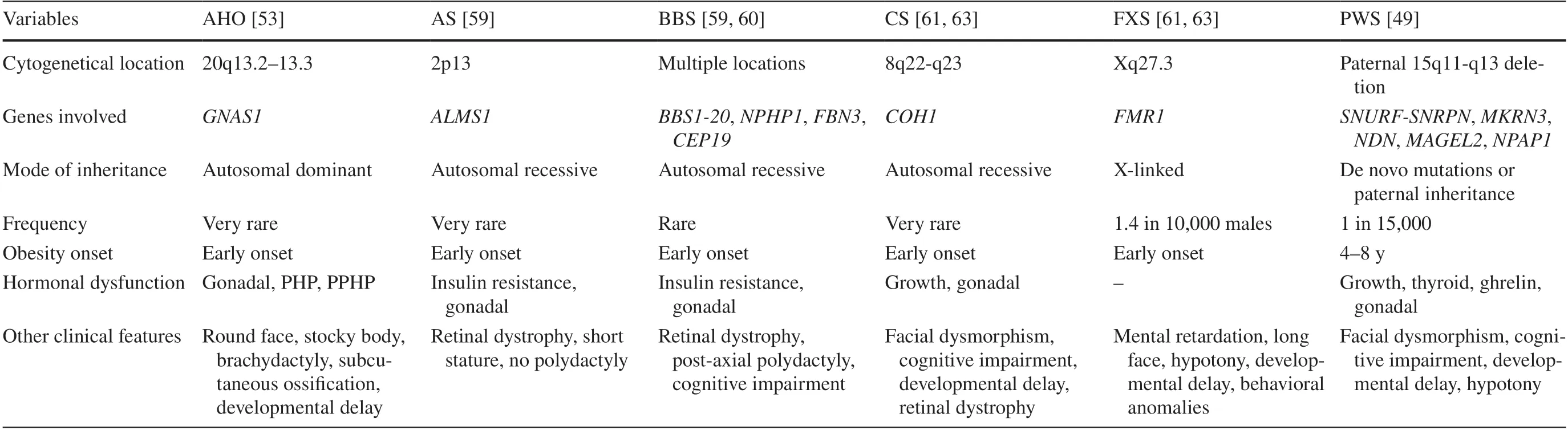
Table 1 Main causes of syndromic obesity
Monogenic obesity
Leptin deficiency is an extremely rare condition that is caused by mutations in the leptin gene [23, 77].Heterozygous mutations are associated with severe hyperphagia and early-onset obesity after normal birth weight, and homozygous mutations, which are extremely rare, are also associated with frequent infections, hypogonadotropic hypogonadism,and mild hypothyroidism [78, 79].
Mutations in the leptin receptor gene (LEPR) are somewhat more common than congenital leptin deficiency, producing similar manifestations in the presence of adequate or elevated leptin levels, being found in up to 3% of patients with severe early-onset obesity, without association with delayed psychomotor development [80, 81].They also present alterations in growth hormone and thyroid function[82].
Mutations inMC4Rare the most common cause of monogenic obesity, which causes 2%–6% of cases of extreme childhood-onset obesity [38, 83].Those with homozygous mutations have higher levels of obesity and hyperphagia, are usually taller than their peers and have no dysmorphisms,and those with heterozygous mutations almost always develop obesity and hyperphagia [84].Extreme hyperinsulinemia and low blood pressure are also associated [85].
POMC deficiency syndrome is very rare and results in deficiency of its products, ACTH and MSH [38, 86].In its homozygous form, it manifests with early ACTH deficiency,adrenal insufficiency, prolonged neonatal jaundice, hyperphagia, obesity, skin pallor, and reddish hair, the latter as a result of poor stimulation of melanocytes [23, 87].Heterozygous mutations may not include adrenal insufficiency and other classic manifestations [88].
ThePCSK1andPCSK2genes encode proteases involved in the processing of neuropeptides and prohormones in endocrine tissues [89, 90].Mutations in these genes alter the processing of gastric peptides and proinsulin, which can result in neonatal severe malabsorptive diarrhea, postprandial hypoglycemia, and obesity [91].In addition, they can lead to impaired growth, hypothyroidism, arginine vasopressin deficiency, and hypogonadotropic hypogonadism [92].Impaired POMC processing directly causes hyperphagia and early-onset morbid obesity [89, 93].Mutations in thePCSK1gene are inherited in an autosomal recessive manner, while the mode of inheritance ofPCSK2mutations has not been described [89].
Although extremely rare, mutations in the genes encodingTrkBandBDNFhave also been implicated in thedevelopment of obesity [94, 95].In patients with WAGR syndrome, which includes Wilms’ tumor, aniridia, genitourinary anomalies, and mental retardation, deletions of chromosome 11p responsible for BDNF haploinsufficiency are associated with lower levels of serum BDNF and with childhood-onset obesity [96].
Other mutations that have been discovered through exome and whole-genome sequencing are currently under study,such as mutations in kinase suppressor of Ras 2, tubby-like protein, carboxypeptidase, and retinoic acid-induced genes[38].The main causes of monogenic obesity are described in Table 3.

Table 3 Main causes of monogenic obesity
Polygenic obesity
Polygenic obesity is the most common cause of obesity in childhood and occurs under the influence of both environmental and genetic factors [99–101].It is caused by the combined effects of mutations in multiple genes, each of which has a small isolated effect [resulting in an elevation of body mass index (BMI) of less than 0.5 kg/m2] [99–101].The occurrence of polygenic obesity is characterized by inter-individual heterogeneity, which means that there are differences in the genetic variants present in different individuals [42, 101].
Variants of two genes,MC4Rand the gene associated with fat mass and obesity (FTO), have been identified,and another gene, insulin-induced gene 2, is under study[102–105].On the other hand, there are several phenotypes related to the different genes involved in polygenic obesity,which are thrifty (characterized by lower energy expenditure), adipogenic (increased tendency to store fat), low lipid oxidation (reduced ability to burn fat for energy), sedentary habits, hyperphagic, obesity-related, increased body fat,increased BMI, increased waist circumference, and increased waist-hip ratio [106].These phenotypes are not necessarilyseparate from one another, and it is possible for an individual to display multiple phenotypes at the same time.
Diagnosis
Genetic causes of obesity are not usually considered unless there is developmental delay or dysmorphic features, which is why the Endocrine Society recommends looking for genetic causes in children with extreme obesity with onset before 5 years, hyperphagia, or a family history of extreme obesity.However, the problem is that hyperphagia has not been well defined, making it necessary to establish an operational definition [85].
BMI is recommended for diagnosing overweight and obesity in adolescents and children aged 2 years and older [85].It is calculated by dividing body weight in kilograms by the square of height in meters, although it does not provide information on body composition or fat distribution [107].In adults, the World Health Organization defines obesity as a BMI greater than or equal to 30 kg/m 2 [4, 107, 108].In adolescents and children aged 2 years and older, overweight is defined by a BMI greater than or equal to the 85th percentile but less than the 95th percentile for age, obesity by a BMI greater than or equal to the 95th percentile for age,and extreme obesity if the BMI is greater than or equal to 120% of the 95th percentile for age or greater than or equal to 35 kg/m 2 .In children under 2 years of age, overweight is defined by a weight-for-height greater than or equal to the 85th percentile, and obesity is defined as greater than the 97.7th percentile [85].This is summarized in Table 4.
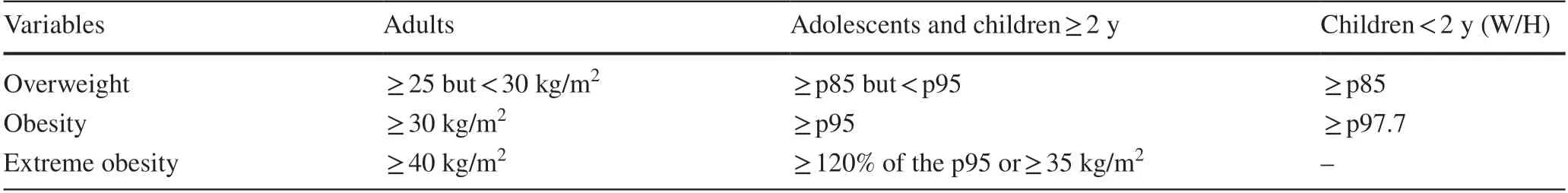
Table 4 Diagnosis of overweight and obesity according to body mass index (World Health Organization)
The presence of manifestations of a syndromic cause of obesity, such as developmental delay, dysmorphic features,and other hormonal deficiencies or vision loss, could guide the diagnostic approach [54].Clinically based diagnostic criteria exist for BBS and PWS [109–113]; however,genetic evaluation is necessary for diagnosis.Initially,microarray,MC4Rgene mutation analysis, and leptin levels should be evaluated [25].Figure 3 presents a diagnostic algorithm with the existing tests.Some of the conditions described do not have commercially available tests.Whole-exome sequencing and next-generation sequencing should be performed to expand the possibility of finding the underlying cause in those where initial testing does not provide definitive results, but there is a high suspicion of a syndromic cause of obesity [25].
Treatment
Obesity is managed at three therapeutic levels: lifestyle modifications, pharmacological treatment, and bariatric surgery [69, 114, 115].The most important part of treatment is lifestyle modification, including dietary counseling and increased physical activity [85].This requires commitment and adequate training of both the patient and their caregivers.Genetic defects can result in changes in energy utilization and storage, and conventional caloric restriction may not be sufficient, necessitating more tailored dietary interventions.For example, in PWS, increased insulin sensitivity and lower resting energy expenditure make it necessary to adopt a low-carbohydrate diet to minimize the risk of obesity [116, 117]; however, during infancy, infants may require support to ensure adequate nutrition and prevent failure to thrive [46].BBS patients can benefit from a hypocaloric diet with restriction of simple sugars and regular aerobic exercise (adapted if blindness is present) [54].Individuals with mutations in theFTOgene can reduce their risk of obesity by simply increasing physical activity [118].
Alterations in the gut microbiota contribute greatly to obesity in children, making it a potentially effective target for management through interventions such as prebiotic or probiotic supplementation or fecal transplantation [119–121].
If limiting weight gain and improving comorbidities have not been achieved after an intensive lifestyle change program, pharmacotherapy for obesity may be initiated[85].Phentermine/topiramate, bupropion/naltrexone, liraglutide, and semaglutide are approved for use only in those older than 16 years with non-syndromic or monogenic obesity, along with a lifestyle modification program, and their use should be discontinued and re-evaluated if there is no greater than 4% reduction in BMI/BMIZscore after use in full doses for at least 12 weeks [85].In a 52 weeks multicenter randomized clinical trial, liraglutide did not show a significant reduction in body weight in children and adolescents with PWS compared to placebo, although it did reduce hyperphagia, requiring further studies to assess its possible benefit in this population [122].
Several drugs are under study for the management of obesity associated with genetic factors.For instance, setmelanotide, an MC4R agonist, has demonstrated a significant reduction in body weight in patients with BBS [123].However,no benefits have been found in patients with PWS, and the results in patients with AS have been inconclusive [123, 124].Livoletide, an inactive ghrelin analog that functions by reducing the level of active ghrelin in the brain, did not demonstrate any benefits in terms of hyperphagia and body weight in patients with PWS [124].Congenital leptin deficiency can be managed with the use of recombinant leptin (metreleptin), a safe and effective treatment for decreasing obesity and improving gonadal and immune function [125–127].Individuals with homozygousLEPRmutations do not benefit from recombinant leptin treatment [38, 128].Obesity due toMC4Rmutations still has no therapeutic options for clinical practice[38].Although there are no therapeutic options for mutations in thePCSK1andPCSK2genes in clinical practice, these are currently being studied [38].Oxytocin is another potential treatment target [129, 130], as well as BDNF [131–134] and drugs with activity on the endocannabinoid system [31, 135,136].Tirzepatide, a dual glucose-dependent insulinotropic polypeptide (GIP) and GLP-1 receptor agonist, and retatrutide, an agonist of the GIP, GLP-1, and glucagon receptors,show promising potential as treatments for obesity.However,currently, there are no studies on their use in obesity caused by genetic factors [126, 137].
Bariatric surgery is safe and effective in adolescents and adults [138–140] and is an option for those who have pubertal development in Tanner stage 4 or 5 and adult size, BMI greater than 40 kg/m 2 or greater than 35 kg/m 2 and extreme comorbidities, family stability, adherence to healthy lifestyles, access to a center with experience in pediatric bariatric surgery, and a multidisciplinary team for subsequent followup [141–146].It should not be performed in children and preadolescents, pregnant or lactating adolescents, those who do not demonstrate adherence to healthy lifestyles, and those who have any untreated psychiatric problems [85, 147, 148].To avoid an unsuccessful operation, preoperative genetic testing of patients with a history of early-onset obesity might be essential, especially considering the potential availability of novel pharmacological treatment options [149, 150].
A systematic review and meta-analysis demonstrated that laparoscopic sleeve gastrectomy (LSG), gastric bypass (GB),and biliopancreatic diversion (BPD) can safely achieve rapid weight loss in patients with PWS [151], with the most significant weight loss occurring during the first year following surgery [152].As high ghrelin levels are responsible for hyperphagia, with 70% of ghrelin being produced by the stomach,procedures that involve partial gastrectomy, such as LSG and BPD, are more likely to result in reduced hyperphagia [153,154].However, observational studies have indicated that the weight loss achieved in these patients is not sustainable in the long term, with results not maintained for periods of up to 5–10 years after the procedure [155, 156].While some level of cognitive impairment may be present in PWS, current guidelines no longer consider this a contraindication for bariatric surgery [147, 157].It is crucial to conduct a careful assessment, involve the family, and provide follow-up in a multidisciplinary setting for these patients [117].
In a study that included 1014 patients undergoing bariatric surgery with a BMI > 50 kg/m 2 and onset of obesity before the age of 10, 3% were found to have genetic obesity caused by heterozygous mutations in theMC4R,POMC, orPCSK1genes.The percentage of body weight loss after GB was not significantly different in patients with genetic mutations compared to those without genetic obesity.Only patients with mutations inMC4Rwho underwent LSG showed less body weight loss compared to those without genetic causes, making GB the preferred surgical option [158].In a retrospective study, patients with obesity and biallelic variants inLEPR,POMC, andMC4Rgenes underwent bariatric surgery at a median age of 19 years.Although the surgery initially led to a median maximum reduction of 21.5 kg in body weight,there was a subsequent median weight regain of 24.1 kg over a maximum follow-up period of 19 years post-surgery [149].
Precision obesity medicine
Precision medicine aims to tailor preventive, diagnostic, and therapeutic approaches to an individual's unique characteristics to enhance disease classification and optimize treatment outcomes by accounting for variability among patients [159].The development of genetic study technologies allows us to approach personalized treatment, considering individual risk profiles so that the therapeutic response is optimal, which includes personalized prescription of diet, physical exercise,and drugs [160].Although this is possible for disorders such as leptin deficiency, in general, it is still far from being a reality, but important progress is being made toward it [161].
Limitations
The small number of patients with rare genetic disorders makes it difficult to collect enough data to draw meaningful conclusions.The cost and complexity of genetic testing make it challenging to identify and diagnose these conditions.Additionally, there may be genetic and environmental factors that interact in complex ways to cause obesity, which makes it difficult to isolate the specific genetic factors that contribute to the condition.Finally, the naming and classification of these syndromes can be inconsistent and confusing, which can make it difficult to communicate about these conditions and conduct research.
Conclusions
The population affected by obesity is increasing exponentially, and obesity with a genetic cause is often underdiagnosed.One of the main reasons for this is that clinicians tend to view obesity as a disease that results from poor lifestyle choices and lack of self-management, and genetic factors are not always assessed.However, new treatments are currently under investigation that could potentially change the prognosis for patients with obesity-associated genetic syndromes.To date, many important aspects of the pathophysiology of obesity remain unknown, and there are several gaps in our understanding of the genetic causes of obesity.Therefore,further research is necessary.
Supplementary InformationThe online version contains supplementary material available at https:// doi.org/ 10.1007/ s12519- 023- 00757-z.
Author contributionsCZMJ contributed to conceptualization, methodology, reviewing and editing, and project administration.QAJE contributed to conceptualization, methodology, investigation, writing of the original draft, and project administration.DVMdC, VdlCJdC,BCCM, and PLSL contributed to investigation and writing of the original draft.GOER contributed to investigation, reviewing and editing.ALEdJ, LCF, ZGFE, and CULA contributed to reviewing and editing.PIJ contributed to conceptualization, methodology, reviewing and editing.All authors approve the final version of the manuscript.
FundingNone.
Data availabilityData sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Declarations
Ethical approvalNot needed.
Conflict of interestNo financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.The authors have no conflict of interest to declare.
 World Journal of Pediatrics2024年1期
World Journal of Pediatrics2024年1期
- World Journal of Pediatrics的其它文章
- Editors
- Information for Readers
- Instructions for Authors
- Current status of Mycoplasma pneumoniae infection in China
- Coinfection of SARS-CoV-2 Omicron variant and other respiratory pathogens in children
- PACS gene family-related neurological diseases: limited genotypes and diverse phenotypes