
PACS gene family-related neurological diseases: limited genotypes and diverse phenotypes
2024-04-14 10:06:42HanZhangKaiGaoShuangWangYueHuaZhangZhiXianYangYeWuYuWuJiang
World Journal of Pediatrics 2024年1期
Han Zhang·Kai Gao,4·Shuang Wang·Yue-Hua Zhang·Zhi-Xian Yang·Ye Wu·Yu-Wu Jiang,4,5
Abstract Background The PACS gene family has been demonstrated to be related to intracellular vesicular trafficking.The phenotypic manifestations caused by the pathogenic variants of PACS include epilepsy, intellectual disability/developmental delay, and malformations, such as facial abnormalities.Methods We identified seven new cases with pathogenic or likely pathogenic PACS variants using next-generation sequencing.Detailed information obtained from these patients was analyzed along with that obtained from previously reported patients.Results With the inclusion of the newly diagnosed cases in this study, 103 cases with PACS gene family-related neurological diseases were reported, of which 43 were PACS2-related cases and the remaining were PACS1-related cases.Most patients had seizures, which have been reported to be effectively controlled by several types of anti-seizure medications (ASMs).The most efficacious and frequently prescribed ASMs included sodium valproate (43.3%, 13/30), oxcarbazepine/carbamazepine(26.7%, 8/30), and levetiracetam (20%, 6/30).Almost all patients had intellectual disability/developmental delay.The most common pathogenic missense variants were PACS1 p.Arg203Trp and PACS2 p.Glu209Lys.In addition, we report a patient carrying a likely pathogenic copy number variation (CNV) (de novo heterozygous deletion of chr14:105821380-106107443,286 kilobase, destroyed part of the furin-binding region domain and the protein structure after it) with more severe and refractory late-onset epilepsy.Conclusions The clinical phenotypes of the different PACS heterozygous missense variants were similar.The pathogenic variant sites of PACS1 and PACS2 were quite limited but located in different regions.A CNV destroying part of the PACS2 gene might also be pathogenic.These findings may provide an important clue for further functional studies on the pathogenic mechanism of neurological disorders related to the PACS gene family.
Keywords Developmental delay·Epilepsy·Intellectual disability·PACS1 ·PACS2
Introduction
ThePACSgene family, which has been demonstrated to be involved in intracellular vesicular trafficking, organellar communication and gene expression [1–3], includes two highly homologous genes in humans,PACS1andPACS2.Variants of both genes are reportedly associated with neurological disorders.The pathogenic variants ofPACS2are associated with developmental and epileptic encephalopathy type 66 (OMIM #618067).Since 2018 [4], 10 studies have reported a total of 38 cases ofPACS2-related neurological disease [5–14].Based on the available clinical records,most patients had seizures and intellectual disability (ID)/developmental delay (DD).Some patients also exhibited structural malformations, such as facial abnormalities.The pathogenic variants ofPACS1are associated with Schuurs-Hoeijmakers syndrome (OMIM #615009).Since 2012 [15],59 cases ofPACS1-related neurological diseases have been reported, whose chief presentation included facial abnormalities, ID/DD, seizures, and structural malformations [16,17].Thus, the clinical phenotypes resulting from the pathogenic variants ofPACS1andPACS2were similar, but the pathomechanism remains to be elucidated.In this study, we reported six cases ofPACS-related neurological disorders,including five cases ofPACS2-related neurological disorder and one case ofPACS1-related neurological disorder.In addition, we report that a CNV (copy number variation)(heterozygous deletion of chr14:105821380-106107443,286 kilobase) might cause aPACS2-related disorder.We also collated, summarized, and analyzed all reported cases ofPACSgene family-related neurological diseases and our newly diagnosed cases to elucidate the genotypic and phenotypic characteristics ofPACS-related neurological disease.
Methods
Ethics statement
This study was approved by the Ethical Committee of Peking University First Hospital.The patients’ parents provided written informed consent for the publication of their case details.All data were analyzed anonymously.
Participants
Pathogenic or likely pathogenic variants were identified in seven Chinese patients with epilepsy.All available data,including clinical manifestations, electroencephalography(EEG) findings, brain magnetic resonance imaging (MRI)features, and gene variant information, pertaining to these seven patients and previously reported patients withPACSgene family-related neurological disease were collated.Patients were excluded if they met one of the following conditions: (1) the pathogenicity of the variant was uncertain;(2) the original paper was not written in English or Chinese;and (3) the cases were found in public databases.
Variation analysis
We collected 5 mL of peripheral venous blood from the patients and their parents.Genomic DNA was extracted for trio-based whole-exome sequencing (WES).The pathogenicity of the variants was predicted using Sorting Intolerant from Tolerant, Polymorphism Phenotyping version 2,combined annotation-dependent depletion, and Mutation Taster.Variants were evaluated against the overall population in the 1000 Genomes Project, GnomAD, and ExAC databases.Pathogenicity was classified using variant classification standards based on the 2015 American College of Medical Genetics and Genomics guidelines.Patients with a suspected causative CNV identified by WES also underwent whole genome sequencing (WGS) to identify the extent of the CNV and analyze its pathogenicity.
Results
Phenotypic summary
With the inclusion of the new cases diagnosed in the current study, 43 cases ofPACS2-related neurological diseases and 60 cases ofPACS1-related neurological diseases have been reported to date.Table 1 summarizes the information on allPACS2-related neurological disease cases, except for those with incomplete clinical data.Seto et al.summarized the information of 52 cases ofPACS1-related neurological disease in 2020 [16], and Tenorio-Castano et al.added seven cases in 2021 [17].Epilepsy, ID/DD, and structural abnormalities were the chief manifestations ofPACSgene family-related neurological diseases.
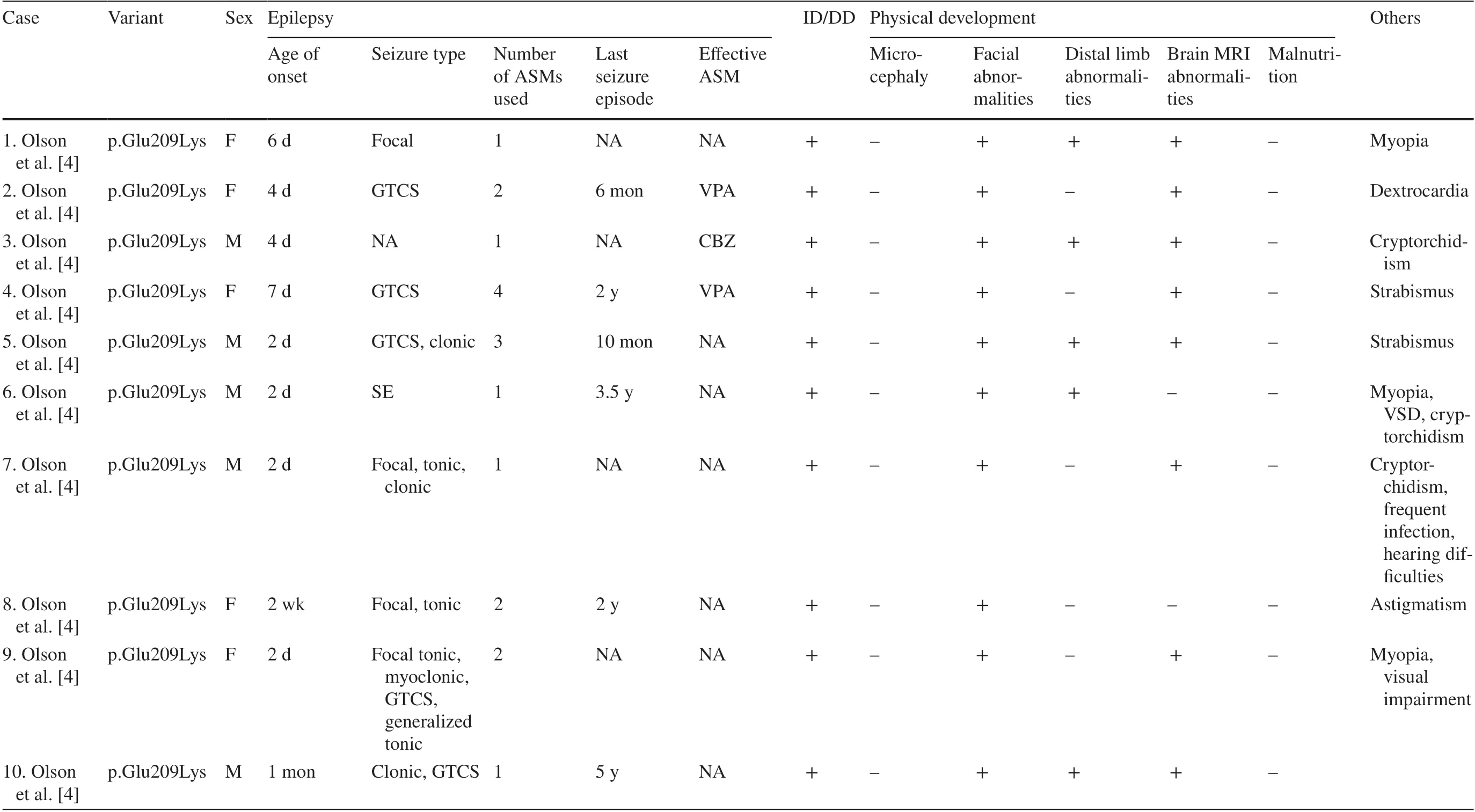
Table 1 Detailed data of 28 patients with PACS2-related neurological diseases
Seizures
Seizures were the characteristic and most frequent first symptom ofPACSgene family-related diseases.All 28PACS2-related patients, whose detailed information was available, experienced seizures as the first symptom,whereas 60% of patients withPACS1-related neurological diseases had seizures [16].Focal seizures were the principal seizure type; 59.4% (19/32) of patients with detailed records had focal seizures.Moreover, tonic seizures, generalized tonic-clonic seizures, clonic seizures, and myoclonic seizures were also observed in some patients.Most patients'seizures were easily controlled by anti-seizure medications(ASMs), and 6/22 patients withPACS2-related neurological diseases (with complete medication data records) had used only one ASM, and 10/22 had used two ASMs.However, detailed records of the seizures were available for only three previously reported patients withPACS1-related neurological diseases, and the follow-up duration was short.In 2018, Martinez-Monseny et al.reported a patient with c.607C>G (p.Arg203Trp) [18], who experienced seizure onset at 3 years of age and presented with focal seizures that were treated with oral carbamazepine.Both patients carrying c.607C>G (p.Arg203Trp) reported by Hoshino et al.in 2019 had seizures [19].Onset of tonic seizures occurred in the neonatal period in one patient, which could be completely controlled by oral phenobarbital; EEG performed during the inter-ictal period showed no abnormalities.In the other patient, seizures developed two days after birth and manifested as apnea, which was completely controlled by oral phenobarbital; the seizures recurred at 2 years and 4 months of age after ASM discontinuation and were treated with oral sodium valproate.In our study, one newly diagnosed patient withPACS1-related neurological disease presented with focal seizures at four days after birth and was treated with phenobarbital and levetiracetam, and no seizures were observed for up to two months.The patient experienced another seizure episode during discontinuation of phenobarbital and dose reduction of levetiracetam.Sodium valproate and levetiracetam were administered four months after birth, and complete control of the seizures was achieved again.The patient had no seizures from 6 months through 9 years of age, i.e., the present day.Several types of ASMs have been reported to be effective for the treatment of epilepsy.The most efficacious and frequently used ASMs were sodium valproate (43.3%, 13/30), oxcarbazepine/carbamazepine (26.7%, 8/30), and levetiracetam (20%, 6/30).Most patients achieved freedom from seizures.ASMs achieved complete seizure control within 6 months of age in 8 of 24(33.3%) patients withPACSgene family-related neurological diseases whose complete epilepsy treatment was available(including case 25, who experienced recurrence at 17 years of age, which was controlled by levetiracetam monotherapy),and complete seizure control was achieved within 2 years of age in 18 of 24 (75%) patients.The patients’ inter-ictal EEG showed slow background rhythm and abnormal wave discharge.Focal seizures of unknown origin were recorded on EEG in case 25.EEG returned to normal in case 5 one month after complete seizure control.
Intellectual disability/developmental delay
Almost all patients (88/89) withPACSfamily gene-related neurological diseases had ID/DD of varying severity.Patients with milder ID/DD (case 25) could graduate from junior high school.At the age of 20 years, this patient could communicate to the bare minimum and imitate others to prepare simple handicrafts.However, his language skills were poor.Patients with more severe ID/DD were likely to have developmental regression and lack self-care ability, language ability, or motor abilities, such as running and jumping.
Structural abnormalities
Height and weight development were normal in patients withPACSfamily gene-related neurological diseases.The most common physical malformations were facial abnormalities, as observed in 96.6% (86/89) of patients, which were characterized by orbital hypertelorism, narrow palpebral fissures, low and flat nasal bridges, spherical nasal tips,low-set ears, thin lips, and micrognathia.Some patients had microcephaly and dysplasia of the distal limbs (fingers and toes).Abnormal neuroimaging findings were observed in most patients: 60.7% (17/28) of patients withPACS2-related neurological diseases exhibited abnormal findings on brain MRI, which included widening of the subarachnoid space,cerebellar dysplasia, and dysplasia of the corpus callosum.However, patients withPACS1-related neurological diseases often present with cerebellar vermis dysplasia on brain MRI.Several patients had other malformations.Cardiac structural abnormalities [16], such as atrial septal defect (ASD), bicuspid aortic valve, aortic constriction, aorto-pulmonary dilatation, aortic valve dysplasia, pulmonary valve dysplasia,patent ductus arteriosus, patent foramen ovale, persistent left superior vena cava, and ventricular septal defect (VSD),were observed in 42% of patients withPACS1-related neurological diseases; 21% had skull deformities or microcephaly;33% had ocular abnormalities, which manifested as astigmatism, strabismus, myopia, hyperopia, and optic disk irregularity; and 7.7% had structural abnormalities of the urinary tract, which manifested as duplex kidney, extra-renal pelvis,and posterior urethral compartment.Some patients had other structural abnormalities, such as cryptorchidism, congenital diaphragmatic hernia, and pulmonary dysplasia.Abnormalities have also been reported inPACS2-related diseases.The patients could present with cardiovascular abnormalities,such as ASD, VSD, and dextrocardia and ocular disorders,such as myopia, strabismus, and astigmatism.
Genotype summary
Forty-three of the 103 reported patients withPACSgene family-related neurological diseases carried pathogenic variants ofPACS2, while 60 carried pathogenic variants ofPACS1.Forty-two of 43 (97.7%) patients withPACS2-related neurological diseases carried a missense variant,c.625G>A (p.Glu209Lys), 1/43 patient (case 28) carried a missense variant, c.607C>G (p.Glu211Lys).A total of 59/60 (98.3%) patients withPACS1-related neurological diseases carried the same type of missense variant c.607C>G(p.Arg203Trp), and one carried another missense variant c.608G>A (p.Arg203Gln).
The sites of pathogenic variation in the PACS1 and PACS2 proteins were quite localized (Fig.1).The pathogenic variants ofPACS1were missense variants involving Arg203.Arg203 was located in the furin-binding region(FBR), and missense variation in this amino acid may have affected the recognition or binding between the PACS1 protein and the cargo.The pathogenic point variants ofPACS2were also missense variants, resulting in the substitution of the acidic amino acid glutamate for the basic amino acid lysine.The variant amino acids were located in the middle region (MR), which plays a major regulatory role.The variant amino acids were all adjacent to the auto-regulatory area of the MR, with Glu209 being the last amino acid in the auto-regulatory region.

Fig.1 Comparison of PACS1 and PACS2 protein structures and variant sites.ARR atrophin-1-related region, FBR furin-binding region, MR middle region, CTR C-terminal region
Patient carrying chr14:105821380-106107443 heterozygous deletion
Only one patient with a de novo heterozygous deletion was identified.The clinical phenotype of this patient differed considerably from that of the other patients.The disease history was characterized by late onset.The patient had essentially normal development until 1 year of age without seizures.DD occurred at approximately 1 year of age, and focal and tonic seizures developed from the ages of 1 year and 5 months.The severity of seizures was greater, status epilepticus was a common occurrence, and the administration of sedative drugs was needed.The patient had refractory epilepsy and had used four ASMs.No seizure episodes occurred for up to one year during the administration of oxcarbazepine and levetiracetam.The patient experienced seizures one month before the last follow-up, and drug titration was underway to obtain the optimal pharmacotherapeutic regimen.The patient's facial abnormalities were similar to those of other patients withPACSgene family-related neurological diseases.
Our study is the first to report that a heterozygous deletion, chr14:105821380-106107443 (286 kilobase), might be associated with the pathogenesis ofPACS-related neurological disease.Trio-WES and CNV sequencing revealed that he carried this de novo CNV, and we performed WGS, which confirmed that he carried a heterozygous deletion measuring 286 kilobases in length (chr14:105821380-106107443)(Fig.2 a).This fragment containedPACS2,C14orf80,CRIP1,CRIP2,IGHA2,IGHE,IGHG4,MTA1,TEX22,andTMEM121.This CNV destroyed part of the FBR and the protein structure after it (Fig.2 b).This heterozygous deletion has not been reported in the Database of Genomic Variants.Karyotype analysis yielded normal results.Trio-WES and WGS did not reveal any other pathogenic variants associated with this phenotype.
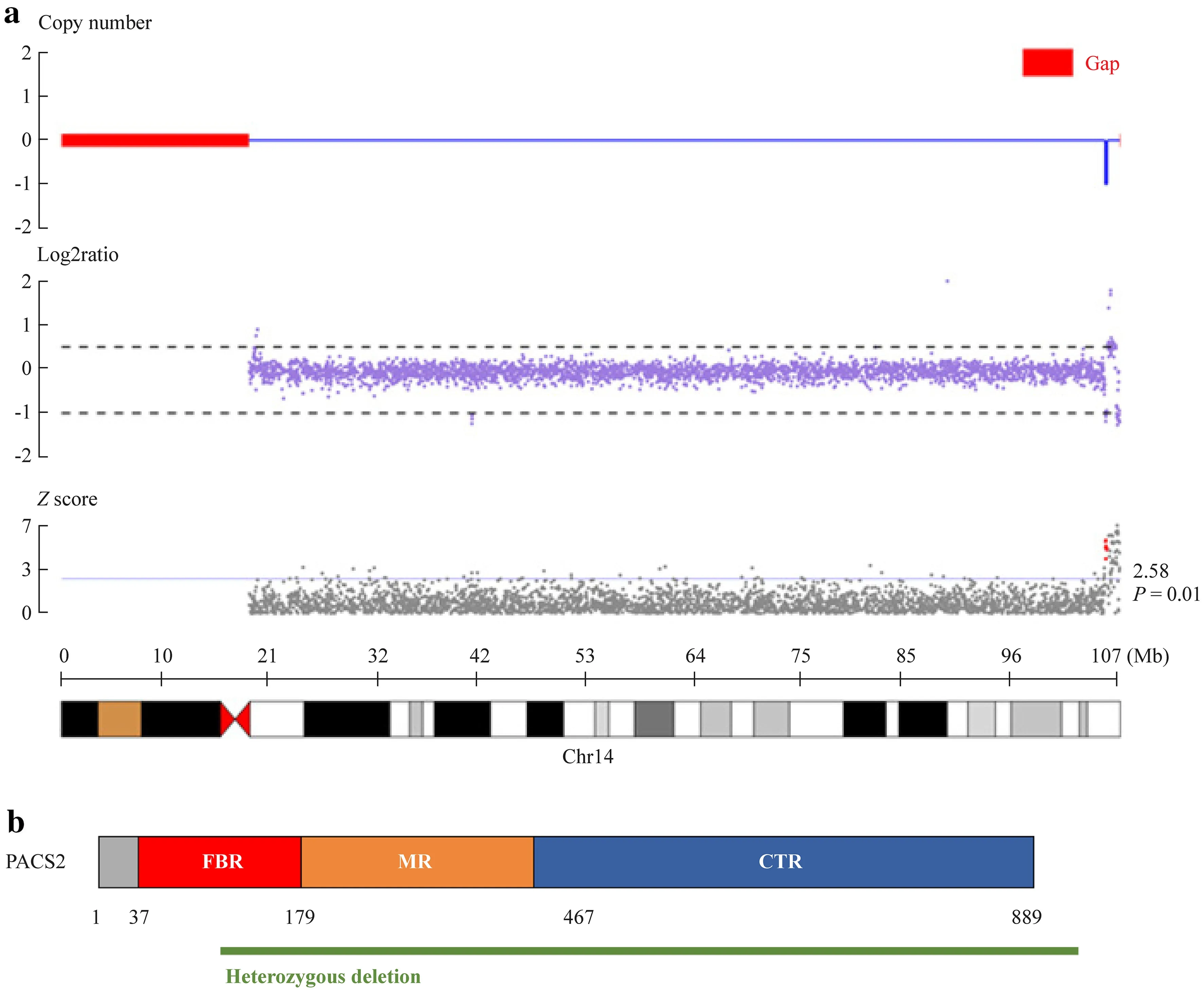
Fig.2 The heterozygous deletion we reported.a WGS detected a heterozygous deletion of chr14:105821380-106107443 (286.06 kilobase); b this CNV caused deletion of a partial segment of PACS2,including half of the FBR and the latter.WGS whole genome sequencing, CNV copy number variation, FBR furin-binding region,MR middle region, CTR C-terminal region
Discussion
In humans, thePACSfamily includes thePACS1andPACS2genes.The pathogenic variants of both genes are associated with neurological diseases with similar clinical manifestations, chief of which are epilepsy, ID/DD of varying severity, and structural abnormalities.Epilepsy is the most common first symptom, which mainly presents as focal seizures and could be associated with a variety of seizure types and status epilepticus.Seizures are easily controlled in most patients, and seizure freedom can be achieved by ASMs in infancy.The most efficacious and frequently used ASMs were sodium valproate, oxcarbazepine/carbamazepine, and levetiracetam.In these cases, the choice of ASM was usually based on the type of seizure.Broad-spectrum ASMs, such as sodium valproate and levetiracetam, are commonly used.Several cases were characterized by focal seizures and were therefore treated with oxcarbazepine/carbamazepine.The choice of pharmacotherapy is usually not based on the causative gene becausePACS-related seizures are usually easy to control.Some patients had started to use effective ASMs before the causative gene was identified.Almost all patients had facial abnormalities, and some even had abnormal brain MRI findings, microcephaly, and multiple visceral structural malformations.However, the phenotype of the patient with heterozygous deletion of chr14:105821380-106107443 diagnosed in our study differed from that of patients with otherPACS2-related neurological diseases.This patient had more severe and refractory epilepsy of later onset, in addition to frequent episodes of status epilepticus.The facial abnormalities and microcephaly observed in this patient were similar to those of the other patients.
PACS1andPACS2are highly homologous [1], and both are derived from the prevertebratePACSgene.Both genes have been implicated in intracellular vesicular trafficking[20].PACS1 localizes cargo to the trans-Golgi network,whereas PACS2 is involved in vesicular transport from the Golgi to the endoplasmic reticulum (ER) and ER to the Golgi, cell membranes, mitochondria, and other pathways.Currently, PACS2 is also considered to be related to apoptosis, autophagy and other pathways [1–3].PACS1 and PACS2(also called PACS-1 and PACS-2) both contain three regions[20], viz.the FBR, MR, and C-terminal region (CTR).The FBR, which is the functional domain, possesses the highest degree of homology and is involved in recognition and binding between the proteins and the cargo.The MR contains nuclear localization signals and phosphorylation sites, which play a regulatory role in the function of the PACS, while the specific function of CTR remains unclear.The two prevalent pathogenic variants ofPACS1were missense mutations involving Arg203.Arg203 is located in the FBR, and the missense variation in this amino acid may have affected recognition or binding between the PACS1 protein and the cargo.Both variants affected the same CpG dinucleotide.Studies in zebrafish have suggested a dominant-negative or gain-of-function patho-mechanism inPACS1-related neurological disorders [15, 21].The two pathogenic point variants inPACS2were one amino acid apart; both involved switching of glutamate to lysine, resulting in remarkably similar clinical phenotypes, suggesting a similar pathogenic mechanism.Both variants were located in the MR, i.e., the regulatory domain, with Glu209 being the last amino acid in the auto-regulatory region.The presence of two indels in this region suggests that the structure near this site may be highly relevant for the regulatory function of PACS2.Co-immunoprecipitation analysis showed that the PACS2 p.Glu209Lys reduced the reversibility of FBR binding to client proteins, which may disturb cellular function [4].This finding indicated that, in contrast toPACS1, the missense variants ofPACS2may be loss-of-function.On the other hand,the loss-of-function constraint measure ofPACS2was 0.279 according to the GnomAD database, which suggested that thePACS2gene is less resistant to loss-of-function variants.
We reported a patient with a heterozygous deletion of chr14:105821380-106107443.This fragment containedPACS2,C14orf80,CRIP1,CRIP2,IGHA2,IGHE,IGHG4,MTA1,TEX22, andTMEM121; however, none of the other genes, exceptPACS2, has been reported to be associated with epilepsy or ID/DD, and onlyPACS2,CRIP1, andMTA1are expressed in the central nervous system.This CNV destroyed part of the FBR domain and the protein structure after it, which might significantly affect protein function, and therefore, we believe that this variant causes neurological disease mainly through its effect onPACS2.This microdeletion might be loss-of-function-like missense variants.However, more functional studies are needed.In 2012, Holder Jr et al.reported one patient carrying a 0.31-Mb microdeletion of chromosome 14q32.33 who presented with epilepsy, DD,and facial dysmorphism [22].However, the pathogenicity could not be determined because they only performed CGH array and fragile X testing, and they did not analyze the segregation of the microdeletion in the family.The clinical phenotype of the patient with heterozygous deletion of chr14:105821380-106107443 diagnosed in our study may be different from that of patients with otherPACS2-related neurological diseases.Does this suggest that PACS2 protein dose dependence in humans is related to developmental period? These findings may provide clues to elucidate the pathogenic mechanism and the precise treatment ofPACS2-related neurological diseases, although they require subsequent functional research.However, a patient with a heterozygous deletion of chr14:105821380-106107443 experienced later onset of epilepsy and severe disease.Hence, the possible pathogenic contribution of other genes in the fragment showing high expression in the nervous system, includingCRIP1andMTA1, to the epileptic phenotypes or refractory mechanisms still cannot be discounted.
In conclusion, this study reported one newly diagnosed patient withPACS1and five patients withPACS2-related neurological disease in a Chinese population and reported that the heterozygous deletion of chr14:105821380-106107443 might causePACS2-related disorders.We summarized the clinical phenotypes and genotypes of the cases diagnosed over the course of this study and those of previously reported cases and found that epilepsy and structural changes associated with heterozygous missense variants in allPACSfamily genes were similar.Patients carrying heterozygous deletions in thePACS2gene have a more severe neurological phenotype than those with otherPACS2-related neurological diseases.The pathogenic mutation sites in thePACSfamily genes are quite limited, but the functional domains where the common mutations ofPACS1andPACS2genes are located are different, viz.the FBR (the main functional domain) and MR (the regulatory domain), respectively.However, we could not explain the mechanism underlying the variations in the severity of the clinical phenotypes and high concentrations ofPACS1andPACS2pathogenic variants in patients carrying the samePACS2pathogenic variants.This suggests a need for further investigation with a larger sample size and further analysis into the genetic background, relationship between phenotype and environment,and biological function.Our findings have provided an important cornerstone for further functional studies and research on the pathogenic mechanisms of associated neurological diseases, in addition to useful clues for future precision treatment studies.
AcknowledgementsThe authors thank the patients mentioned in the article and their families for the clinical information they provided and their contribution to scientific research.
Author contributionsZH and GK contributed equally to this manuscript and they were co-first authors.ZH contributed to study design, collection, evaluation and interpretation of the data,and drafting and writing of the manuscript.WS, ZYH, YZX, and WY contributed to collection, evaluation and interpretation of the data.GK and JYW contributed to study design, collection, evaluation and interpretation of the data, and revised the manuscript and approved the final version.
FundingThis work was supported by the National Key Research and Development Program of China (No.2020YFA0804000), the National Natural Science Foundation of China (Nos.81971211, 12026606, and 81601131), Key Project of Clinical Medicine Research of National Clinical Research Center for Child Health and Disorders, Children’s Hospital of Chongqing Medical University (No.NCRCCHD-2021-KP-02), Beijing Natural Science Foundation (No.7212109), the Beijing Key Laboratory of Molecular Diagnosis and Study on Pediatric Genetic Diseases (No.BZ0317), the Capital Health Research and Development of Special Fund (No.2020–1-4071), National High Level Hospital Clinical Research Funding (Scientific Research Seed Fund of Peking University First Hospital) (No.2022SF29), and the Fundamental Research Funds for the Central Universities (Nos.BMU2017JI002,BMU2018XY006, and PKU2017LCX06).The funding agencies had no role in the study design, the experiments, analysis, or interpretation of data, the writing of the report, or the decision to submit the article for publication.
Data availabilityMost data generated or analyzed during this study are included in this published article.Other data are available from the corresponding author on reasonable request.
Declarations
Ethical approvalThis study was approved by the Ethical Committee of Peking University First Hospital.The individuals’ parents in this manuscript had given written informed consent to publish these cases details.All data were analyzed anonymously.
Conflict of interestAuthor Yu-Wu Jiang is a member of Editorial Board forWorld Journal of Pediatrics.The paper was handled by the other Editor and has undergone rigorous peer review process.Author Yu-Wu Jiang was not involved in the journal's review of, or decisions related to this manuscript.All authors declared no financial or nonfinancial conflict of interests related to this paper.
 World Journal of Pediatrics2024年1期
World Journal of Pediatrics2024年1期
- World Journal of Pediatrics的其它文章
- Editors
- Information for Readers
- Instructions for Authors
- Current status of Mycoplasma pneumoniae infection in China
- Coinfection of SARS-CoV-2 Omicron variant and other respiratory pathogens in children
- Neighborhood predictors of short sleep duration and bedtime irregularity among children in the United States: results from the 2019–2020 National Survey of Children’s Health